


R包pheatmap绘图(数据Z-scor标准化)
6 个月前
· 来自专栏
表观遗传学研究生

路虽远,行则必至;事虽难,做则必成。
现有一组不同组织中的表达量数据,要观察这10个基因更倾向于在哪种组织中高表达,可以通过绘制热图进行观察:
#原始数据(FPKM值)
| Tissue1 | Tissue2 | Tissue3 | Tissue4 | Tissue5 | Tissue6 | |
| Gene1 | 27.192 | 56.238 | 58.092 | 7.416 | 27.81 | 25.956 |
| Gene2 | 20.394 | 56.238 | 26.574 | 3.09 | 21.012 | 4.326 |
| Gene3 | 38.316 | 30.282 | 53.766 | 61.182 | 40.17 | 0.618 |
| Gene4 | 12.36 | 0.618 | 50.676 | 19.776 | 14.214 | 32.136 |
| Gene5 | 53.766 | 9.888 | 33.99 | 37.698 | 4.326 | 35.844 |
| Gene6 | 32.136 | 16.686 | 25.956 | 58.092 | 56.238 | 2.472 |
| Gene7 | 11.742 | 19.776 | 61.8 | 12.36 | 3.708 | 44.496 |
| Gene8 | 59.328 | 47.586 | 50.058 | 47.586 | 0 | 8.034 |
| Gene9 | 14.214 | 35.844 | 31.518 | 27.192 | 59.946 | 6.798 |
| Gene10 | 25.338 | 21.63 | 49.44 | 14.832 | 47.586 | 15.45 |
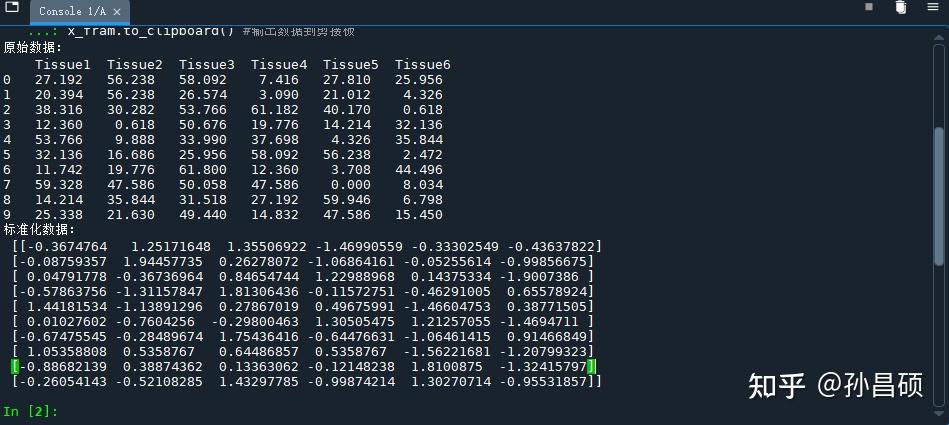
#可以看到数值间的差异很大,为了方便比较,需要进行标准化,因为是比较一个基因的不同组织,所以以行数据进行标准化,使用python进行数据标准化:
import pandas as pd
from sklearn.preprocessing import scale
X = pd.read_clipboard() #将剪切板的数据导入,带表列名无行名的数据
X #输出确认列名及行数
x_scale=scale(X=X,axis=1,with_mean=True,with_std=True,copy=True,)
#Z-socre标注化,计算各个数据与行数据均值的差异,axis=0,列标准化,axis=1,对行标准化,默认0
print('原始数据:\n',X)
print('标准化数据:\n',x_scale)
x_fram=pd.DataFrame(x_scale) #数据转化为表格
x_fram.to_clipboard() #输出数据到剪接板
#将标准化之后的数据在Excel种加上行名和列名,然后用pheatmap绘图:

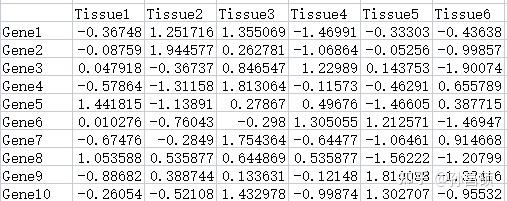
data<- read.csv(file ="clipboard", sep="\t",header = TRUE, row.names = 1) #从剪切板中导入数据
library(pheatmap)
bk=c(seq(-2,-0.1,by=0.01),seq(0,2,by=0.01)) #预设标尺值
pheatmap(data,
cluster_rows = TRUE, #对行进行聚类
cluster_cols = FALSE, #对列进行聚类
clustering_distance_cols = "correlation", #Pearson相关性进行聚类,默认是“euclidean”方式
show_rownames=TRUE, #显示行名
show_colnames = TRUE, #显示列名
scale="none", #不对数据进行标准化
fontsize_row = 10, #行字体大小
fontsize_col = 20, #列字体大小
breaks =bk, #标尺赋值