

ORF与CDS的区别与联系


1.ORF
ORF 的英文展开是 open reading frame(开放阅读框)。
ORF 是理论上的氨基酸编码区,一般是在分析基因序列得到的。把基因的mRNA输入到程序种,程序会自动在 DNA 序列中寻找启动子(ATG 或 AUG),然后按每 3 个核酸一组,一直延伸寻找下去,直到碰到终止子(TAA 或 TAG)。此时程序就把这个区域当成 一个ORF 区,认为理论上可以编码一组氨基酸。但问题是,在一个mRNA中寻找 ATG 并不靠谱。因为寻找到的 ATG 很可能是相邻两个密码子的尾和头的混合体。
ATGCAGCGTACTC看上面这个小序列,有三种蛋白质编码组合的可能
(1)ATG | ORF寻找程序会认为这是一个启动子
(2)GCA | 一个普通的序列
(3)TGC | 一个普通的序列
这就是 ORF 三种框架的来源。实际上,DNA 序列可以按六种框架阅读和翻译(每条链三种,对应六种不同的三联密码子)。
那么哪一种是正确的呢?这得结合基因的产物(蛋白质)来进行确定。
tips:
- 一个mRNA可能有很多个ORF
- 不同的ORF长度不一样,有的甚至只有十几个碱基,这很明显就是一个错误的阅读框。
- 我们一般认为最长的哪个是正确的ORF,要真正确定,需要根据蛋白质的序列来查证。
所以,我们说 ORF 只是理论上的编码区,与真实的情景可能并不一样。
2.CDS
CDS 是 Coding sequence 的缩写,是编码一段蛋白产物的序列,是结构基因组学术语。
与开放读码框 ORF 的区别:
- 开放读码框是从一个起始密码子开始到一个终止密码子结束的一段序列;不是所有读码框都能被表达出蛋白产物,或者能表达出占有优势或者能产生生物学功能的蛋白。
- CDS,是编码一段蛋白产物的序列。
- CDS 必定是一个 ORF。但也可能包括很多 ORF。 反之,每个 ORF 不一定都是 CDS。
Open reading frame (ORF) :
a reading frame that does not contain a nucleotide triplet which stops translation before formation of a complete polypeptide.
Coding sequence (CDS) :
The portion of DNA that codes for transcription of messenger RNA。
3. 小结
开放阅读框是基因序列的一部分,包含一段可以编码蛋白的碱基序列。当一个新基因被识别,其 DNA 序列被解读,人们仍旧无法搞清相应的蛋白序列是什麽。这是因为在没有其它信息的前提下,DNA 序列可以按六种阅读框进行解读。 ORF 是指一个基因的起始密码子到终止密码子的密码子组合,中间不被终止密码子打断,是直接可以翻译成目的蛋白的核酸序列,也就是成熟 mRNA 中的 cds,是我们做目的基因表达需要连入载体的部分。
ORF 识别包括检测这六个阅读框架并决定哪一个包含以启动子和终止子为界限的 DNA 序列而其内部不包含启动子或密码子,符合这些条件的序列有可能对应一个真正的单一的基因产物。ORF 的识别是证明一个新的 DNA 序列为特定的蛋白质编码基因的部分或全部的先决条件。
4.附录: ORF寻找工具
做完转录组后,我们会获得很多基因的转录本序列,如何分析这些转录本序列的开放阅读框(ORF),然后找到对应的蛋白呢?今天就为大家介绍一下使用频率最高的软件,NCBI 的 ORF Finder,以及另一款可以进行批量分析的软件 getORF。
1.ORF Finder
ORF Finder 是 NCBI 开发的一个在线分析转录本序列 ORF 的工具,与 NCBI 整合地很好。在百度里面搜索即可得到:

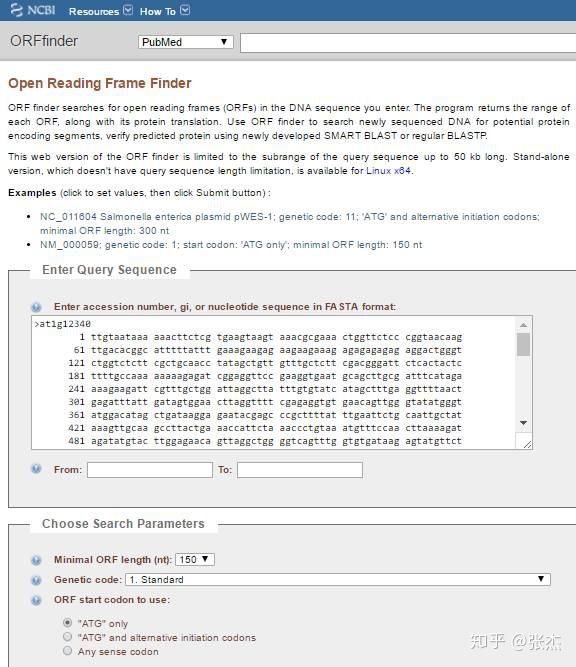
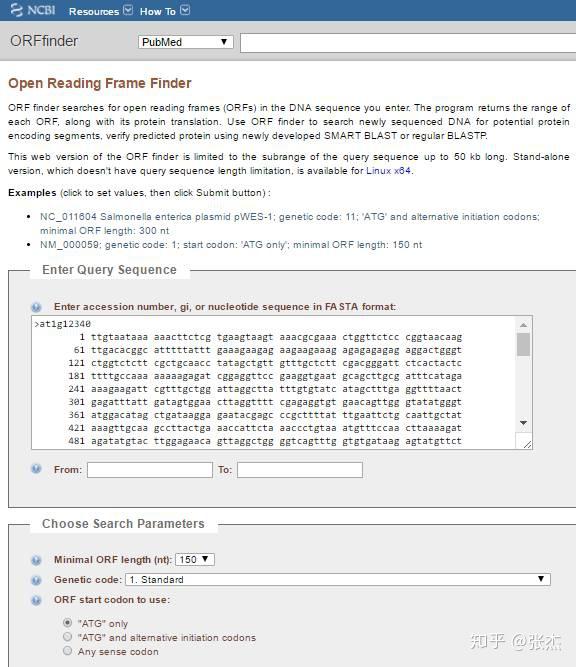
点击第一个或者第二个条目,即进入 ORF Finder 的页面,如下。在搜索框中输入一段转录本序列,也可以写 NCBI 的 GI 号码。在搜索参数部分,还可以设置最小 ORF 序列长度,遗传密码子一般就选择 standard,当然对于原核生物,也可以点开下拉框找到对应的物种类别。起始密码子 start codon 一般选择 ATG only,如果想得到尽可能多的 ORF 也可以选择第三个选项,这样得到的 ORF 就不是以 ATG 开头了。
最终得到的 ORF 序列以图形的形式展现,可以很直观地比较 ORF 的位置和长度,如下:
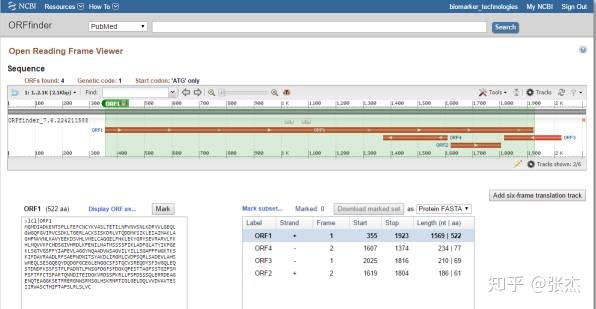
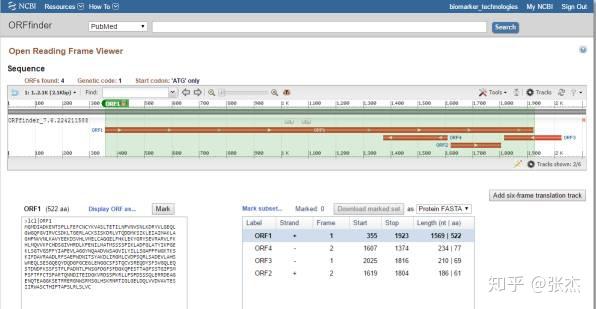
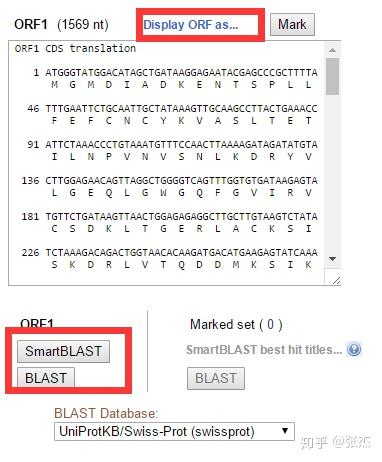
点击不同的 ORF 即可给出相应的氨基酸序列,也可以设置为显示 DNA 序列或者 DNA 和 protein 相互比较的序列形式,如下图标红框的部位。接下来的问题是,如何知道预测出来的氨基酸序列是否可靠呢?一般最长的氨基酸序列就是比较可靠了,也可以直接点击 BLAST 对得到的氨基酸序列进行 blastp 分析,看能否比对到已知的蛋白序列上。
点评
NCBI 的 ORF Finder 简单易用,结果的展现形式也很直观,还能直接对得到的氨基酸序列进行 blastp 分析,以验证 ORF 的可靠性。但缺点是不能进行批量处理,只能一个一个转录本序列进行分析,这对于手上握有大量数据的同学,就有点慢了,下面介绍一款可以批量进行 ORF 预测的软件。
2.getORF
getORF 也是一款经典的用于分析转录本序列 ORF 的软件,属于 EMBOSS 软件包。百度搜索一下 getORF,会出现多个条目,是不同的科研单位搭建的在线运行 getORF 的平台,都可以用,小编在这里推荐一个速度比较快的平台
http://
emboss.bioinformatics.nl
/cgi-bin/emboss/getorf
,界面如下:
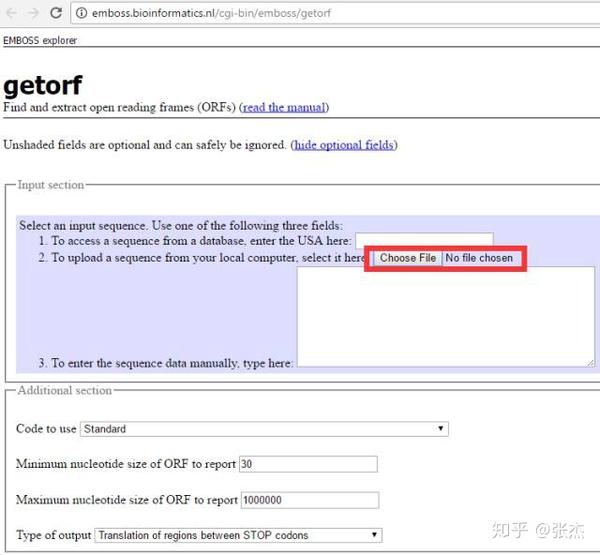
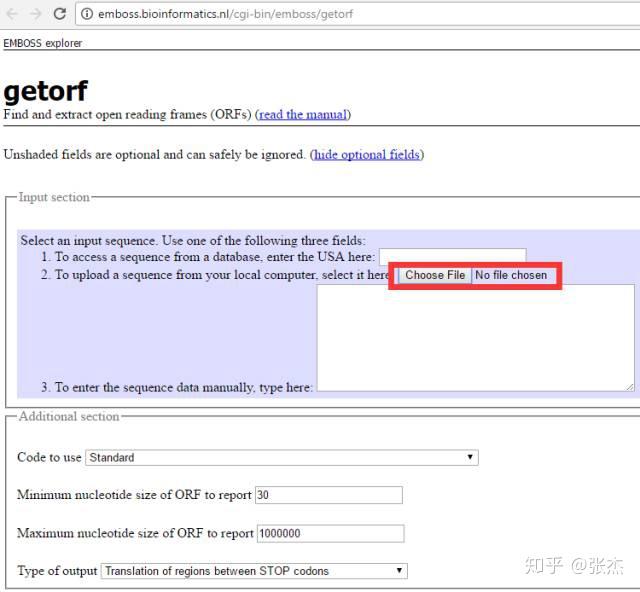
getORF 与 ORF Finder 最大的不同就是可以上传 fasta 格式存储的序列(不知道什么是 fasta 格式的同学请自行百度),这样就可以一次上传很多条转录本序列,批量进行处理。其它的设置与 ORF Finder 相同,可以设置 ORF 长度最大和最小的限制,也可以选择是否以 ATG 开头(Translation of regions between START and STOP codons),默认是不以 ATG 开头(Translation of regions between STOP codons)。
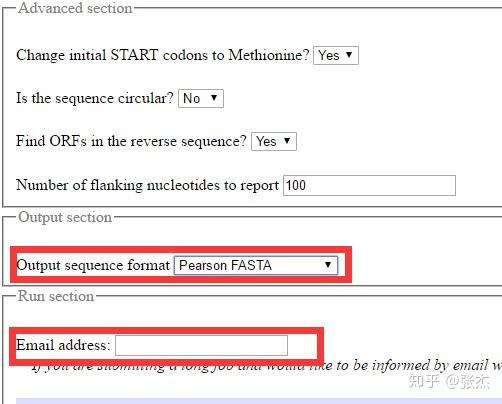

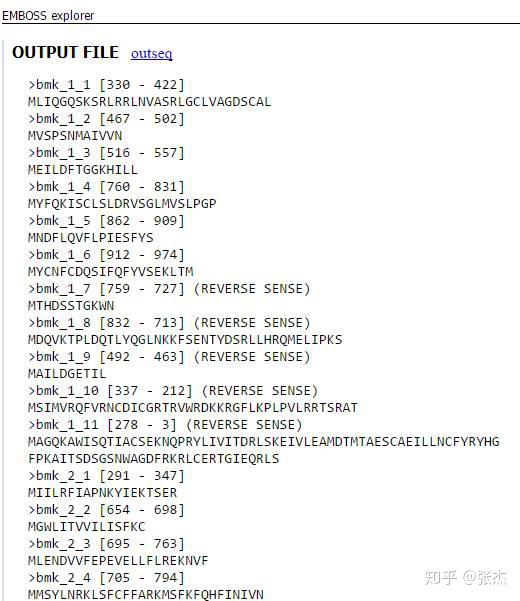
在高级设置里面还可以有多种设置,默认即可。输出的格式也可以有多种选择,还可以填写自己的 email 地址,会发送结果文件的链接到邮箱。最终的输出结果如下图,同一个转录本的不同 ORF 用后缀的数字表示(
1,
2…)。输出结果可以进行复制保存,然后提交到 blastp 进行分析。

点评
getORF 最大的优势是可以进行批量处理,预测得到的 ORF 也很多,但界面的呈现不如 ORF Finder 直观。