


王炸!24小时MS计算免费课程:建模、自由能、过渡态、能带、态密度、光学性质、声子计算、激发态、溶剂化、电子输运、静电势!

Materials Studio (MS)是入门DFT计算的首选软件,其建模功能彪悍,可视化界面简洁方便,并且整合了多功能计算引擎,且无需Linux系统,非常容易上手,备受初学者青睐!
为了帮助大家快速入门DFT计算,拿下顶刊,华算科技杨老师根据MS官方手册,设计了 24小时入门课程,权威靠谱,完全免费 。
内容涉及CASTEP、DMol
3、DFTB+模块,包括表面/吸附建模、能带、态密度、PDOS、Origin作图、弹性常数、声子计算、谱图模拟、差分电荷密度、过渡态搜索、激发态/光学性质、溶剂化效应等。(详见下文)
华算出品,专业靠谱,零基础入门DFT计算,你确定不学?
免费领取: 长按识别下方二维码,公众号后台对话框回复关键词“ MS ”。 获取课程网盘链接、课件、计算工程文件,加入课程群,长期答疑!
长按识别二维码回复:MS
已有超过7000名学员入群,群内提供日常答疑,有任何Materials Studio使用中的问题也可以在课上的答疑环节和课后的群中提问!
课程内容
Materials Studio第一课:CASTEP模块晶格参数预测、能带、态密度、电子密度计算,Origin作图(4小时)
课程内容如下:
1. CASTEP结构优化
2. 能带/态密度/电子密度计算
3. F -43 m 空间群,惯用胞/初基胞转换,周期性镜像
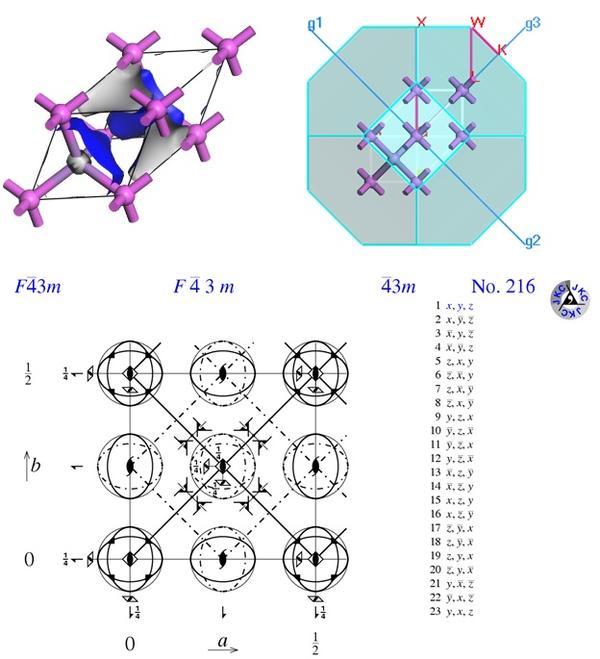

4. 可不可以不做几何结构优化?
5. 泛函选取对结构的影响
6. 几何结构优化不收敛与电子自洽循环不收敛
7. 能带/态密度/PDOS呈现
8. 电子态密度呈现(实操)
9. 怎样用Origin画能带与态密度图(解读)
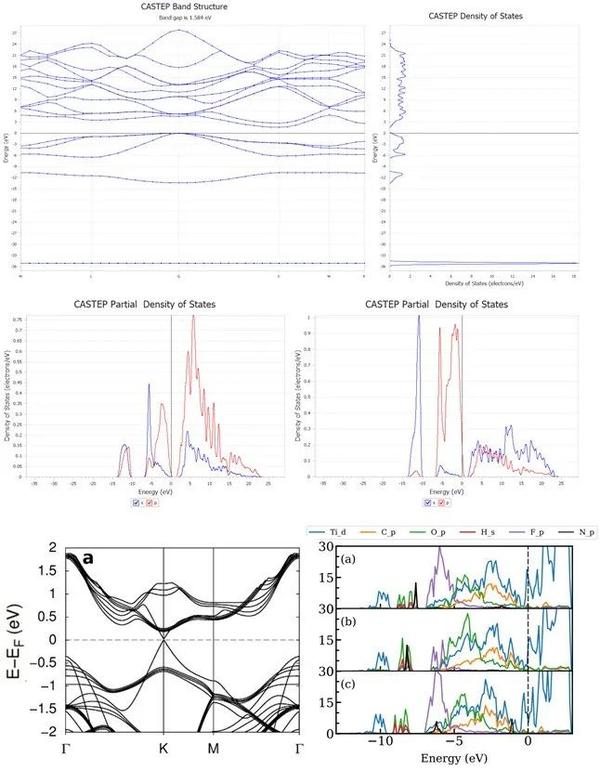

10. 自洽循环与非自洽计算,能带k点路径选取
11. 能带与态密度的意义,如何解读
12. 电子密度等值面的意义
Materials Studio第二课:CASTEP模块表面吸附建模、计算、呈现和分析(含脚本)(4小时)
作为催化计算热点,表面吸附计算一直是大家所关注的焦点之一,如何选取吸附表面,如何建立表面吸附模型,如何对吸附结果进行分析,一直是入门课、零基础班乃至专题班中被问及最多的问题。
杨老师精心选取官方教程中CASTEP模块关于表面吸附计算和性质分析案例,为大家手把手详细解析每步操作及其原因。
课程内容如下:
1. 建立Pd(110)表面模型
2. 建立Pd(110)表面添加CO分子的模型并进行结构优化
3. 吸附能计算与态密度分析
4. 切表面的时候应该切哪个表面?应该切多厚?
5. 表面性质计算时,为什么要固定原子层?
6. 表面吸附计算中,超胞到多大合适?
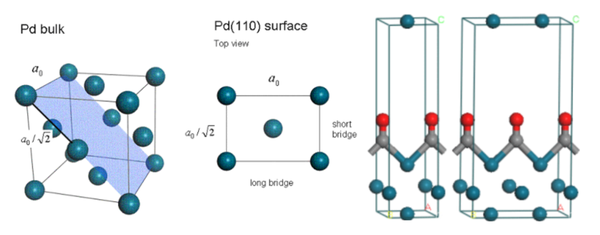

7. 定义原子片段
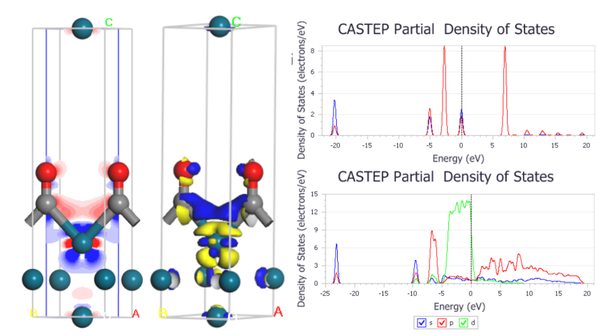
8. Pd(110)表面上的CO分子的电荷差分密度的计算
9. 电荷差分密度图的呈现
10. 电荷差分密度等值面的物理意义
11. 电荷差分密度如何分析电子得失
12. 电荷差分密度图沿z轴的分布及脚本



Materials Studio第三课:CASTEP模块STM图、NMR谱和芯能级谱性质计算分析(4小时)
CASTEP模块中有很多性质可以计算和分析,最常见的如能带和态密度,以及在之前的课程中进行了讲解,力学、光学、热力学等性质,在零基础课中也会涉及。但是Properties选项卡中仍然有零基础班都没有讲到的一些性质,只能看着,不敢尝试?
华算科技精心选取官方教程中CASTEP模块STM图、NMR谱和芯能级谱性质计算分析的教程,为大家手把手详细解析每步操作及其原因,和零基础班、专题班的内容都无重复,送给大家的免费福利,大家还不快来!
由于课程第一部分需要完成Pd(110)表面上CO分子的吸附计算,建议先观看上一期课程视频。
课程内容如下:
1. 模拟Pd(110)表面上CO分子体系的STM图
2. 计算L-丙氨酸17O的NMR谱
3. STM图与DOS、轨道图的联系
4. 如何用模拟的STM图表征表面氧空位,判断吸附位点


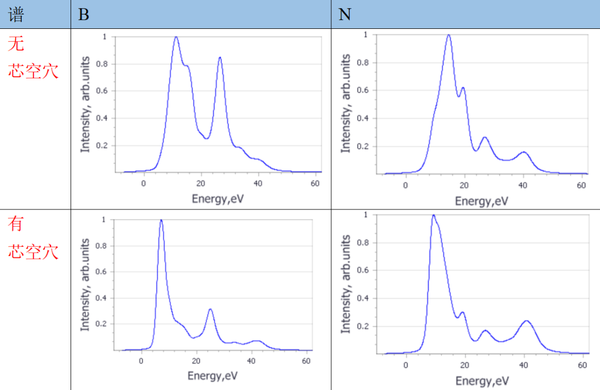
5. 利用第一性原理预测BN的芯能级谱
6. MS中曲线图查看与Origin中处理小技巧
7. 初基胞与惯用胞该如何选(切表面,反铁磁性体系,能带计算时)
8. 为何选择OTFG赝势

Materials Studio第四课: DMol
3模块计算能带结构,态密度,优化结构、激发态/光学性质 (4小时)
江湖上一直流传着“物理人用CASTEP,化学人用DMol
3”的传说,与CASTEP不同,DMol
3不需要三维周期性结构,因此成为了化学专业研究人员研究单个分子、分子之间化学反应和过渡态的利器。
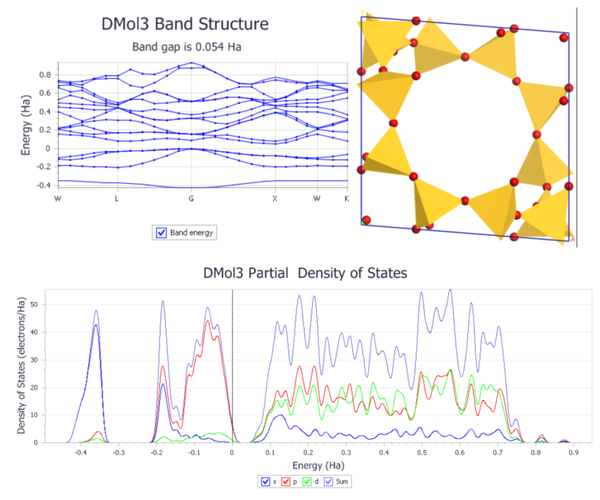
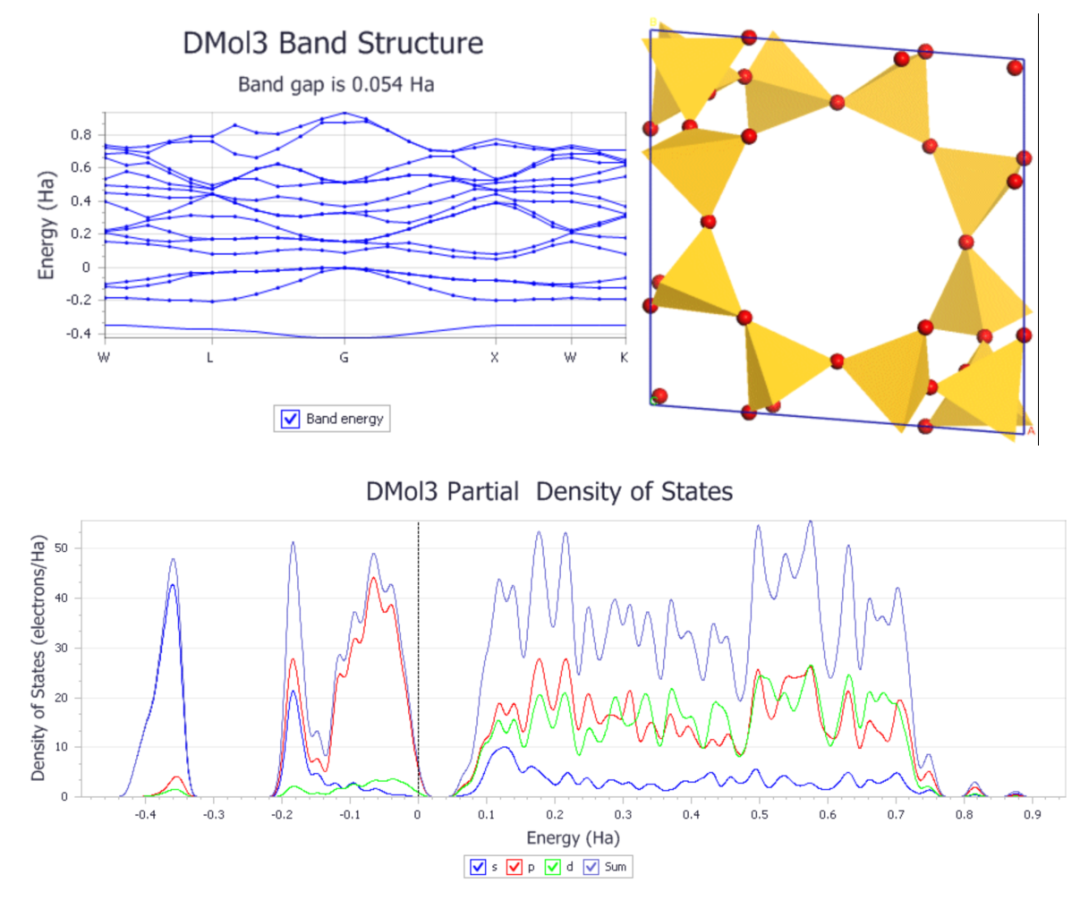
杨老师精心选取官方教程中利用DMol
3计算能带结构和态密度、利用DMol
3中的离域内坐标对固体进行几何优化、计算香豆素的光学性质的教程,基本计算任务、性质计算分析及应用,为大家手把手详细解析每步操作及其原因,帮助大家迅速掌握DMol
3的基础操作。
课程内容如下:
1. 利用DMol
3计算能带结构和态密度
2. 利用DMol
3中的离域内坐标对固体进行几何优化和静电势计算
3.DMol
3和CASTEP方法上的区别,优缺点对比
4.DMol
3的结果文件与计算量
5.DMol
3中的几何结构优化算法
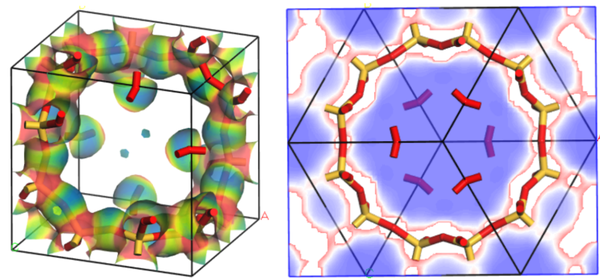
6. 静电势在电子密度等值面投影图的绘制
7. 静电势切面图的绘制
8. 计算香豆素的光学性质
9. 电子密度与静电势的意义
10. 激发态与溶剂化效应


Materials Studio 第五课: DMol
3模块分 析化学反应和过渡态搜索 (4小时)
这一期主要内容是利用DMol
3分析化学反应和过渡态搜索,对化学和材料专业的研究者来说,使用DMol
3进行过渡态搜索和化学反应势垒计算永远是心头的朱砂痣,研究化学反应的小伙伴一定不要错过!
杨老师精心选取官方教程中利用LST/QST工具搜索过渡态、利用DMol
3的LST/QST和NEB工具计算简单化学反应的势垒和Diels-Alder反应的动力学的教程,过渡态、化学反应势垒和反应动力学三大抓人眼球的领域,为大家手把手详细解析每步操作及其原因,帮助大家迅速掌握DMol
3分析化学反应和过渡态搜索的方法。
课程内容如下:
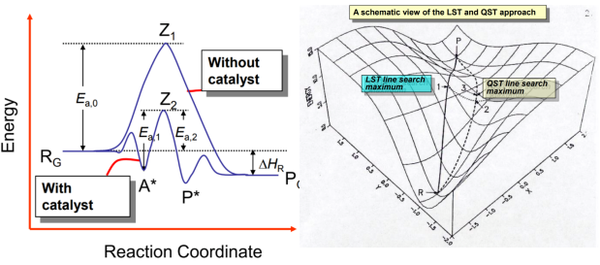
1. 利用LST/QST工具搜索过渡态
2. 利用DMol
3的LST/QST和NEB工具计算简单化学反应的势垒:使用LST/QST计算过渡态
3. 过渡态理论
4. LST/QST和NEB的区别


5. 利用DMol
3的LST/QST和NEB工具计算简单化学反应的势垒:使用TS确认执行NEB计算
6. Diels-Alder反应的动力学
7. 过渡态的虚频验证条件
8. 过渡态确认原理
9. Arrhenius方程与反应速率计算

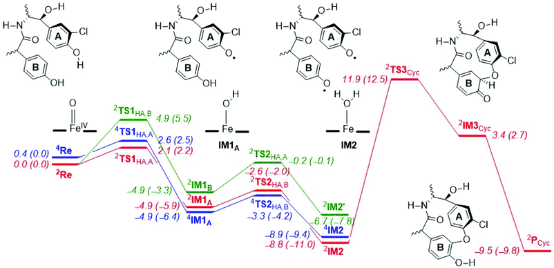

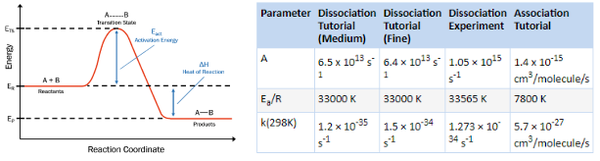
Materials Studio第六课: DMol
3模块官方教学之化学反应与过渡态 (4小时)
本次课程针对DMol
3模块,讲解内容如下:
1. 计算化学反应的自由能
2. 利用DMol
3模拟电子输运
3. 内能,焓,吉布斯自由能的组成
4. 特定温度下振动对吉布斯自由能的贡献
5. 电荷混合参数
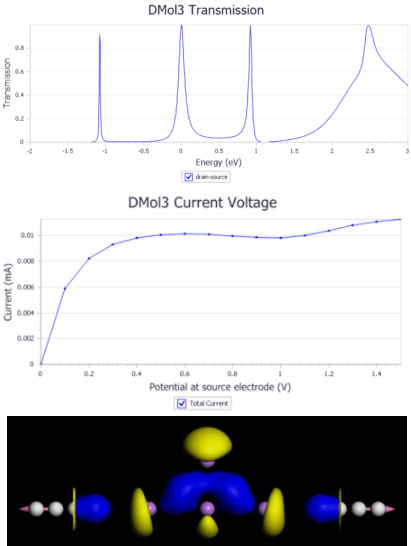
7. 碳纳米管的几何优化(实操)
8. 利用DFTB+模拟电子输运(实操)
9. 非平衡格林函数方法NEGF(解读)
10. DFTB+半经验紧束缚方法(解读)
11. 石墨烯菱形格子变矩形格子,根号三超胞(解读)
12. 在线答疑

亮点直达
1. 本系列入门课程旨在从不同模块入手,带大家逐一重现Materials Studio中各个教程中的操作,精彩不容错过;
2. 结合杨站长多期讲授Materials Studio课程的经验,助大家了解每种操作背后的原因,以使大家将其平移至自己研究的体系;
3. 解读结果分析所涉及的原理,将有助于科学问题分析的结论写进文章中,知其然并知其所以然。
讲师介绍
杨老师(杨站长):华算科技全职技术专家,拥有10年以上Materials Studio软件使用经验。曾就职于德国马克思普朗克研究所,日本WPI研究所,并曾在芬兰阿尔托大学进行长期访问,作为PI主持欧盟与日本科研项目各2项,日本高校科研项目2项。
主要从事固态相变的第一性原理研究、电化学固液界面的AIMD研究与超分子化学中的分子动力学模拟。
王老师(Ravi老师):华算科技兼职技术专家,博士毕业于哈尔滨工业大学,研究方向主要为表界面模拟和晶体生长。在Materials Studio官方教程翻译及教学、文献拆解系列撰写、MS系列教程编辑方面拥有7年经验。
上课方式
免费领取: 长按识别下方二维码,公众号后台对话框回复关键词“ MS ”,获取课程视频、课件、计算工程文件,并加入课程群。
长按识别二维码回复:MS
本次课程由深圳华算科技有限公司、深圳浦华系统技术有限公司联合制作!各模块教程持续更新中,敬请关注!
免费