


新药I期、II期、III期之临床试验设计路径

来源见水印。来源见水印。来源见水印。
新药临床试验设计路径:I期临床试验
1 背景知识
临床试验,英文为clinical trial,而不是clinical experiment。学药童鞋肯定或多或少做过各种实验,不管是化学实验、生物实验还是制剂实验、药理实验,用的都是“实验”二字。所谓“实验”,《现代汉语词典》的定义是为了检验某种科学理论或假设而进行某种操作或从事某种活动;而“试验”是为了察看某事的结果或某物的性能而从事某种活动。而临床试验正是为了考察新药对目标患者的效用而展开的一系列的试验,本系列文章就和大家讲讲新药的临床试验。
新药研发过程主要包括苗头分子的发现、细胞活性评价、非临床药理毒理研究、临床试验和上市后的安全性监督。其中临床试验耗资/耗时基本占整个新药开发的60%~80%,可谓是新药开发最耗钱耗时的阶段。FDA将临床研究分为三期,这三期临床研究通常按期依次实施,但也可以有重叠;另外针对新药批准上市后的临床研究,FDA定义其为IV期临床试验。ICHE8根据临床研究的目的将临床试验同样分为4期。
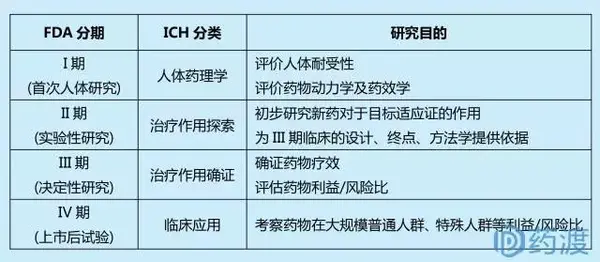
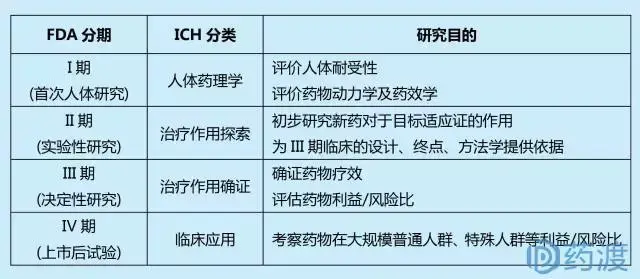
2006年,FDA发布了《Guidance for industry, investigators and reviewers exploratory IND studies》,提出0期临床试验概念,其受试者更少(≤10例),研究周期更短(≤7天),用于探索新药在人体的药代动力学和药效学研究,为I期临床提供指导。不管是0期临床试验概念,还是传统三分法临床试验,其分期的目的是基于风险控制的哲学观。I期临床通常采用少量(数十人)健康志愿者作为受试者,相较于采用较多患者作为受试者的II期以及更大样本量的III期临床试验而言,具有成本低(人少),时间短(可供的健康受试者众多)和低风险(健康受试者身体抵抗力较强)等特点。
2 I期临床试验全景
2.1 是什么(Who)
I期临床试验是在动物药理毒理试验基本成功的基础上,首次应用在人体上,用来初步评价新药的人体耐受性和药代动力学试验。
2.2 为什么(Why)
I期临床试验主要目的是观察随人体给药剂量增加而出现副作用的情况,新药在人体药代动力学性质以及收集有效性的早期证据。通过I期临床试验,观察人体对新药的耐受程度、药代动力学和药效动力学,探索药物最大耐受剂量(Maximal Tolerable Dose,MTD)、剂量限制性毒性(Dose-Limiting Toxicity,DLT),为制定接下来II、III期临床试验设计和给药方案提供依据。
2.3 做什么(What)
I期临床主要回答以下两个问题:①药物的不良反应是什么;③药物是如何被吸收代谢的。为回答以上问题,I期临床至少需要完成以下关键研究:
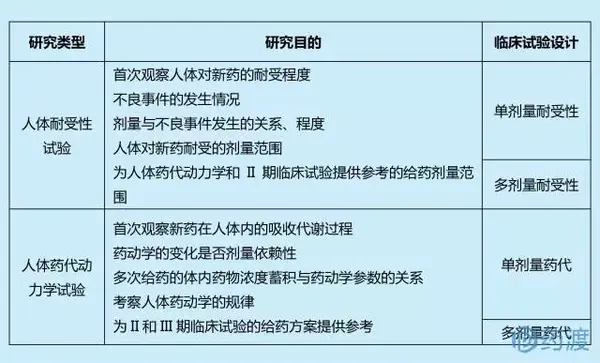
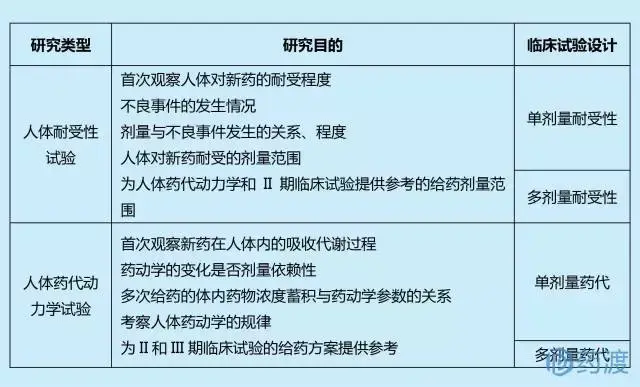
另外,I期临床也可以同步考察新药的人体药效学、食物影响、药物间相互作用等。
2.4 怎么做(How)
试验顺序: 通常依次进行耐受性单剂量试验、药动学单剂量试验、多剂量耐受性和药动学试验。对于采用患者进行的I期临床试验,人体耐受性试验和药动学试验可同步进行。
设计原理: I期临床试验常采用开放、自身对照试验。但当主要不良反应缺乏客观指标或不宜判定不良反应与药物关系时,常采用随机盲法、安慰剂对照试验。
受试者: 多选用健康年轻的男性作为志愿者。但类似细胞毒药物等(如抗肿瘤药物)应采用患者作为受试者。
样本量: 一般为20-80名受试者。
接下来主要介绍I期临床试验设计路径。
3 I期临床人体耐受性试验设计
人体耐受性试验是考察人体对药物不同剂量的耐受程度,通过试验发现出现不良反应性质和剂量。I期临床的人体耐受性试验一般先进行单剂量的探索,在此基础上确定是否进行多剂量试验。试验可以是开放、基线对照,也可以采用随机化和盲法提高观察结果的准确性。I期临床人体耐受性试验总体设计理念:从起始剂量到最大剂量之间设若干组,各个试验组的剂量由小到大逐组进行,直至找到最大耐受剂量(MTD)或到达设计的最大剂量。
3.1 起始剂量
起始剂量是指从动物实验过渡到人体试验时,首次应用于人体的药物剂量。为确定Ⅰ期临床试验的安全起始剂量,需要充分了解临床前动物的药理学、毒理学及药动学数据。确定起始剂量的方法主要有以下5种。
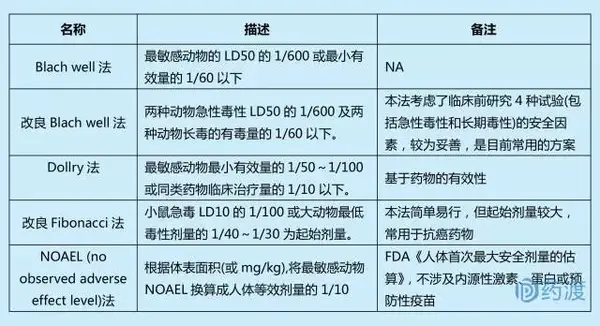
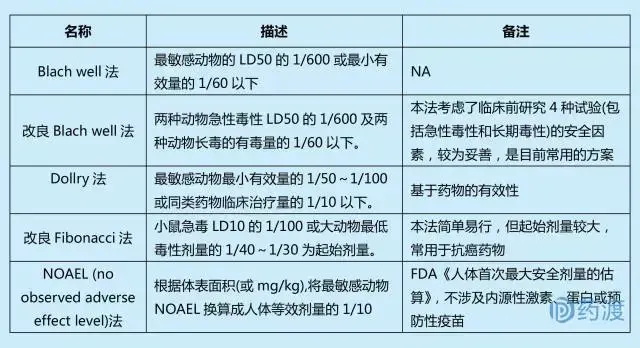
起始剂量的选择应遵循两大原则:①避免毒性反应;②能够快速达到I期临床人体耐受性试验的评估目标。
3.2 最大剂量
最大设计剂量通常有以下三种方法①同一药物、同类药物或结构相似药物单次最大剂量;②动物长期毒性试验中引起中毒症状或脏器出现可逆性变化的剂量的1/10;③动物长期毒性试验中最大耐受量的1/5~1/2。最大剂量范围内应包括预期的有效剂量。
如试验达到最大剂量受试者仍无不良反应时,试验即可结束。剂量递增到出现终止指标或其他较严重的不良反应时,虽未达到最大剂量,也应终止试验。
3.3 剂量递增设计(爬坡试验)
在确定了起始剂量和最大剂量后,需要设计剂量递增方案,以便开展剂量递增的爬坡试验。剂量递增方案的确定要考虑起始剂量与药效学有效剂量和毒性剂量之间的距离、毒代和药代动力学特征等因素。通常采用费氏递增法(改良的Fibonacci法)设计剂量爬坡方案,即当初试剂量为n (g/m2)时,其后按顺序递增的剂量分别是2n、3.3n、5n、7n,此后则依次递增前一剂量的1/3。其特点是开始递增速度快,后期增速较慢,在确保受试者安全的情况下,以合理的速度和梯度迅速达到耐受性临床试验的终止目标。
另外剂量递增设计还有固定比例递增法,即剂量按照固定比例递增,但临床实际应用较少。


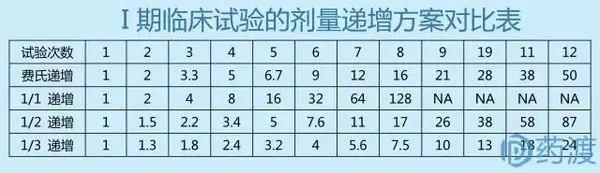
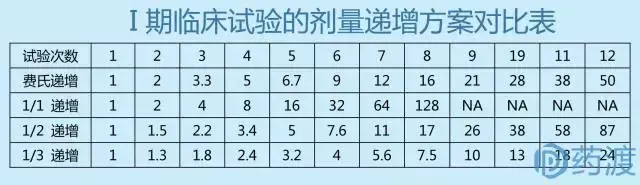
对于剂量递增,也可根据具体药物的自身特点,设计更具针对性的剂量递增方案。剂量递增的基本原则:初期递增幅度可较大,后期递增幅度应较小。递增系数过小,会增加不必要的受试者例数;递增系数过大,会增加受试者的危险性。安全性大或毒性小的药物剂量递增幅度可大,有的可成倍递增;安全性小或毒性较大的药物剂量递增幅度应小。
3.4 最大耐受剂量(MTD)试验设计
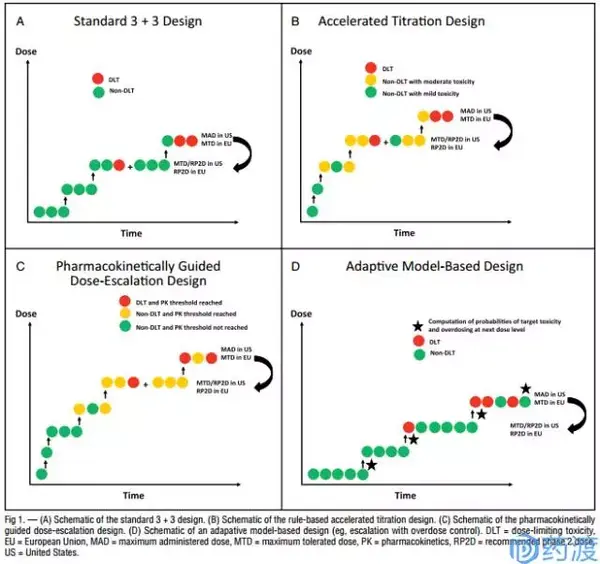
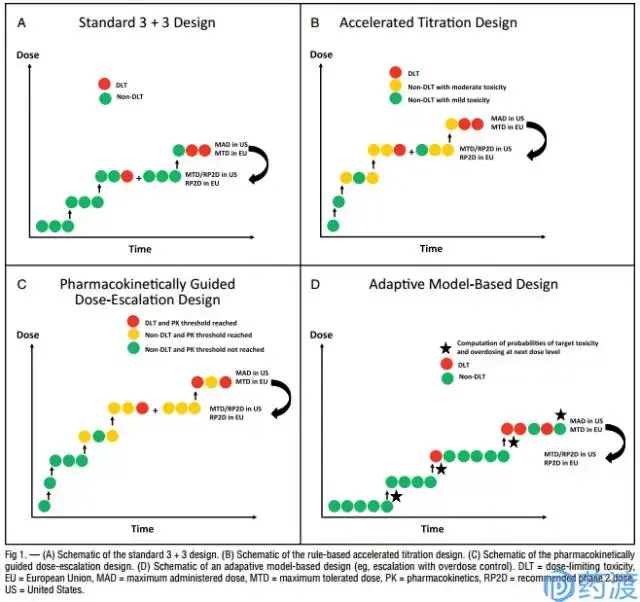
I期临床人体耐受性试验的目标是确定人体的最大耐受剂量。在给定起始剂量、最大剂量和递增剂量的前提下,我们通过设计试验方案来确定人体的最大耐受剂量。确定最大耐受剂量的方法一般有两大类:基于规则的试验设计(Rule-Based Designs)和基于模型的设计(Model-Based Design)。
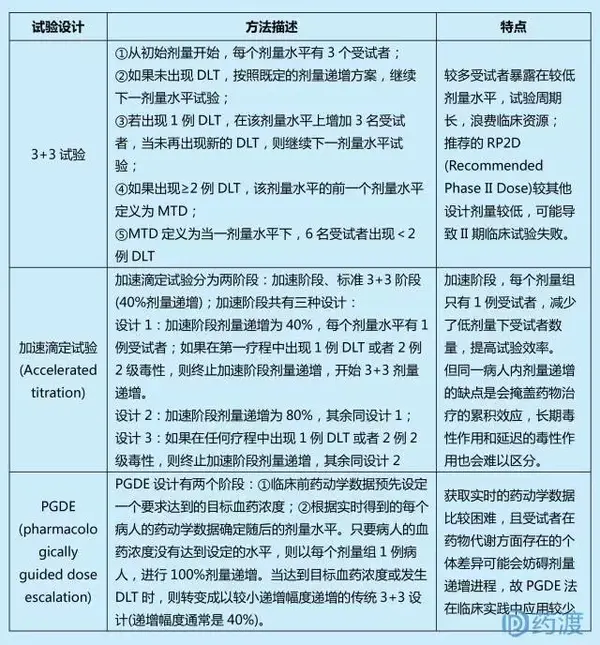
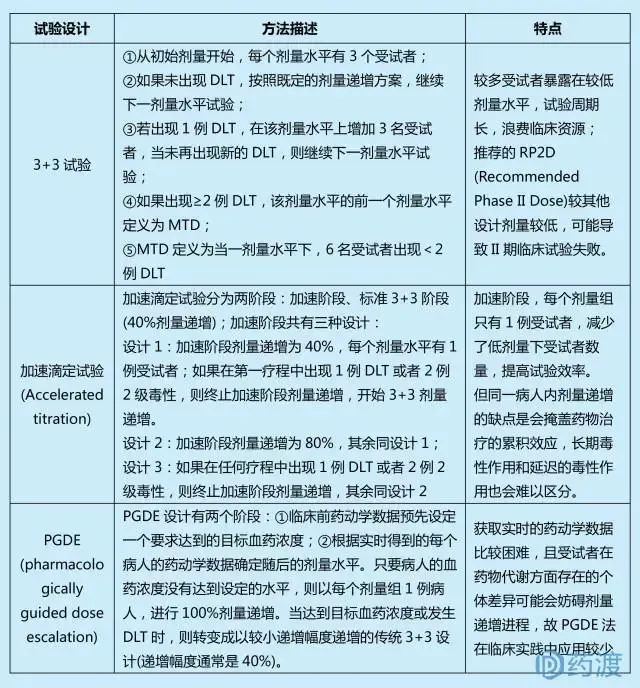
A:基于规则的试验设计(Rule-Based Designs)
关键点:未预先估计剂量-毒性曲线;剂量递增基于当前剂量水平的毒性结果;包括传统的3+3、加速滴定、PGDE试验等。
B:基于模型的设计(Model-Based Design)
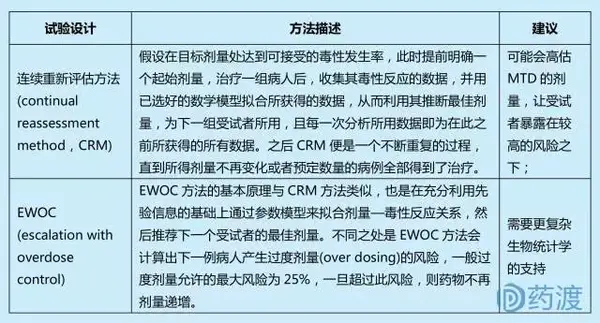
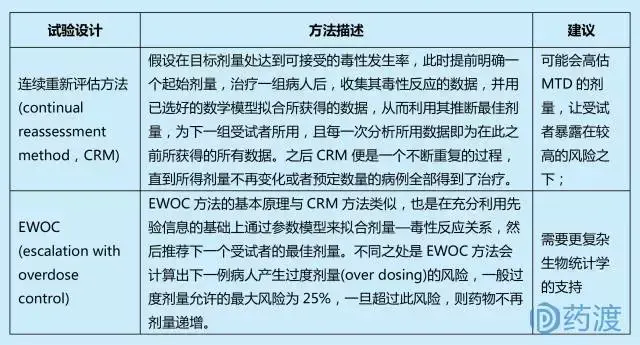
关键点:先于受试者入组前建立剂量-毒性曲线模型;试验过程中,利用受试者毒理数据实时修正剂量-毒性曲线;要求良好的生物统计学支持建立和修正剂量-毒性曲线;基于模型设计包括:CRM、Modified CRM、EWOC、TITE-CRM、mTPI、Mixed-effect POM、Fractional dose-finding试验设计等
4 I期临床人体药代动力学试验设计
I期临床人体药代动力学试验主要目的是评价药物清除率、预测药物或其代谢物在人体可能的积聚、潜在的药物间相互作用等。I期临床人体药代动力学试验一般在人体耐受性试验之后进行。
4.1 试验设计
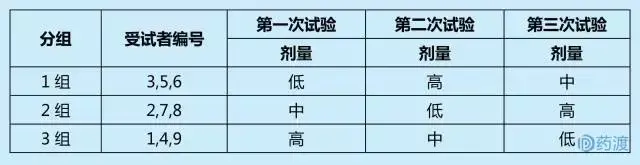
I期临床人体药代动力学试验设计一般为随机交叉自身对照,以减少不同试验周期和个体差异对试验结果的影响。目前主要有两种试验设计:2剂量双周期交叉试验设计、3剂量3周期3×3拉丁方交叉试验设计。以下以3×3拉丁方交叉试验设计为例。
试验基本要求:受试者:9名健康男性。分组:随机分为三组,每组3人。剂量:低、中、高三剂量;高剂量必须接近或等于MTD,中剂量应与临床拟单次剂量接近或相同,三个剂量之间应呈等比或等差关系,以便观察不同剂量-血药浓度是否呈线性关系。两次试验间隔周期:≥7个半衰期
试验设计方案:

另外,药代动力学试验可考虑增加如下试验:
预实验: 在正式进行药代动力学试验前,可考虑进行2-4人的预实验,以便发现可能出现的问题,并为后期的试验条件、剂量大小、观察时间、取样频率等提供参考,以期准确反应药物在人体内药代动力学过程。
食物的影响: 口服固体制剂的药代动力学试验需考察餐前、餐后药动学的差异,尤其是食物对药物吸收程度的影响。
5 多剂量人体耐受性和药代动力学试验
在单剂量人体耐受性和药代动力学试验后,需进行多次给药的耐受性和药代动力学试验,以便考察新药多次给药的人体耐受性以及是否存在药物蓄积作用等。试验基本要求:受试者:一般选择8-12名健康受试者。剂量:基于II期临床试验拟定剂量范围选择1-3个剂量。给药周期:基于单剂量药代动力学中半衰期。
6 思考
I期临床试验是新药首次应用于人体,I期临床的主要目的是考察健康受试者对新药的耐受性和药代动力学性质,为接下来应用于患者的II期临床试验提供治疗剂量和可能出现的副反应提供参考。虽然I期临床试验在新药整个临床试验周期中占比较小,但所谓“欲事之无繁,则必劳于始而逸于终”,一个良好的I期临床,能够尽可能发现新药的特性,为接下来的II、III期临床试验设计提供扎实的事实依据。
参考文献:
1、0 期临床试验:新药临床研究的新模式
2、Accelerated Titration Designs for Phase I Clinical
3、Phase 1 Trial Design Is 3 + 3 the Best ?
4、ICH E8.临床研究的一般考虑
5、关于Ⅰ期临床试验的研究综述
6、FDA: 人体首剂最大安全起始剂量的估算
7、抗肿瘤药物贝叶斯Ⅰ期临床试验设计方法比较研究
8、药物临床试验研究1期
9、创新药物I期临床试验方案设计探讨
10、药物临床试验方法学
11、临床试验的设计与分析
12、新药I期临床试验剂量的探索与确定
13、抗肿瘤药物Ⅰ期临床试验中剂量探索的设计方法概述
新药临床试验设计路径:II期临床试验
1 序言
在I期临床试验明确了药物人体耐受性、安全性、药代动力学特点和推荐的PR2D (Recommended Phase II Dose),即可以开始启动II期临床试验。II期临床试验又称为探索性临床试验,是新药首次在患者身上进行、以探索有效性为目的临床试验。由于新药临床研究费用高昂、周期较长,作为承上启下的II期临床试验设计至关重要。申办者希望通过II期临床试验,能尽快发现很有前景的新药而不至于过早终止研究,同时又希望能尽早终止无效新药的进一步试验。
2 II期临床试验全景图
I期试验侧重于新药安全性和药代动力学,而II期试验重点则转移到药效学上。II期临床试验主要目标是初步考察新药能否在目标患者人群中建立药效学相对比较一致、药物的毒性可以接受的水平。
2.1 是什么(Who)
II期临床试验主要用于评价新药在目标患者上的初步有效性和安全性。
2.2 为什么(Why)
II期临床试验目的与研究药物有效性有关,其寻求回答的问题包括:①药物在I期临床确证的安全剂量范围内对某一特定适应症的有效性如何?②患者短期的新药不良反应和风险是什么?
2.3 做什么(What)
II期临床试验包括:①确定新药作用于目标患者的最大和最小有效剂量范围,为III期临床试验剂量提供参考;②新药产生疗效的血药浓度与药效学参数的关系,即药代动力学和药效学关系。根据目的不同,II期临床有时又分为IIa期和IIb期。


2.4 怎么做(How)
试验顺序: 一般按照IIa期、IIb期序贯实施;
设计原理: II期作为探索性试验,可以采用多种设计方法,如同期对照、自身对照、开放试验、三臂试验(阳性药、安慰剂、试验药)、剂量-效应关系等的研究;
受试者: 目标适应症患者
样本量: 几十到数百人
判断终点: 客观缓解率等
接下来以肿瘤新药的II期临床为例,介绍常见II期临床试验设计。
3 肿瘤新药的II期临床设计
Ⅱ期临床试验设计方法根据有无对照组设置,分为单臂试验和随机对照试验。另外,还包括随机撤药试验设计等。
3.1 单臂临床试验设计
单臂研究(Single Arm Study):即单组临床试验,顾名思义,就是仅有一个组的研究,没有为试验组设计相对应的对照组;常用于新药研发的Ⅱa期。肿瘤新药II期临床试验中,往往要对多个瘤种、多种剂量或用法进行探讨,目的是淘汰无效剂量、筛选敏感瘤种,以便进一步深入研究。单臂试验又分为单臂单阶段和单臂多阶段,最简单的试验设计即为单臂单阶段试验,其在计划的样本数量的病人都接受治疗后,根据治疗效果最后得出试验结论。单臂单阶段试验设计的缺陷是:如果在达到最后样本量之前,发现治疗无效,也不能终止试验,造成资源浪费和伦理学困境。
单臂多阶段试验设计能够避免单阶段试验设计的缺陷,其能在某试验组疗效未达到预期效果时,终止该试验组的研究,避免更多的受试者接受无效治疗。
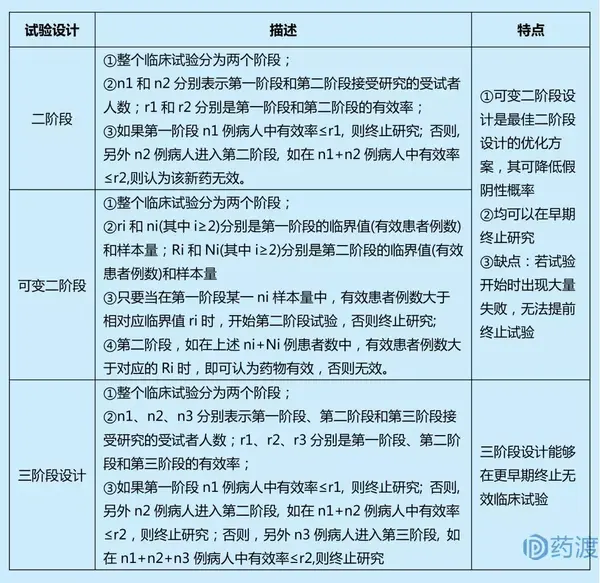
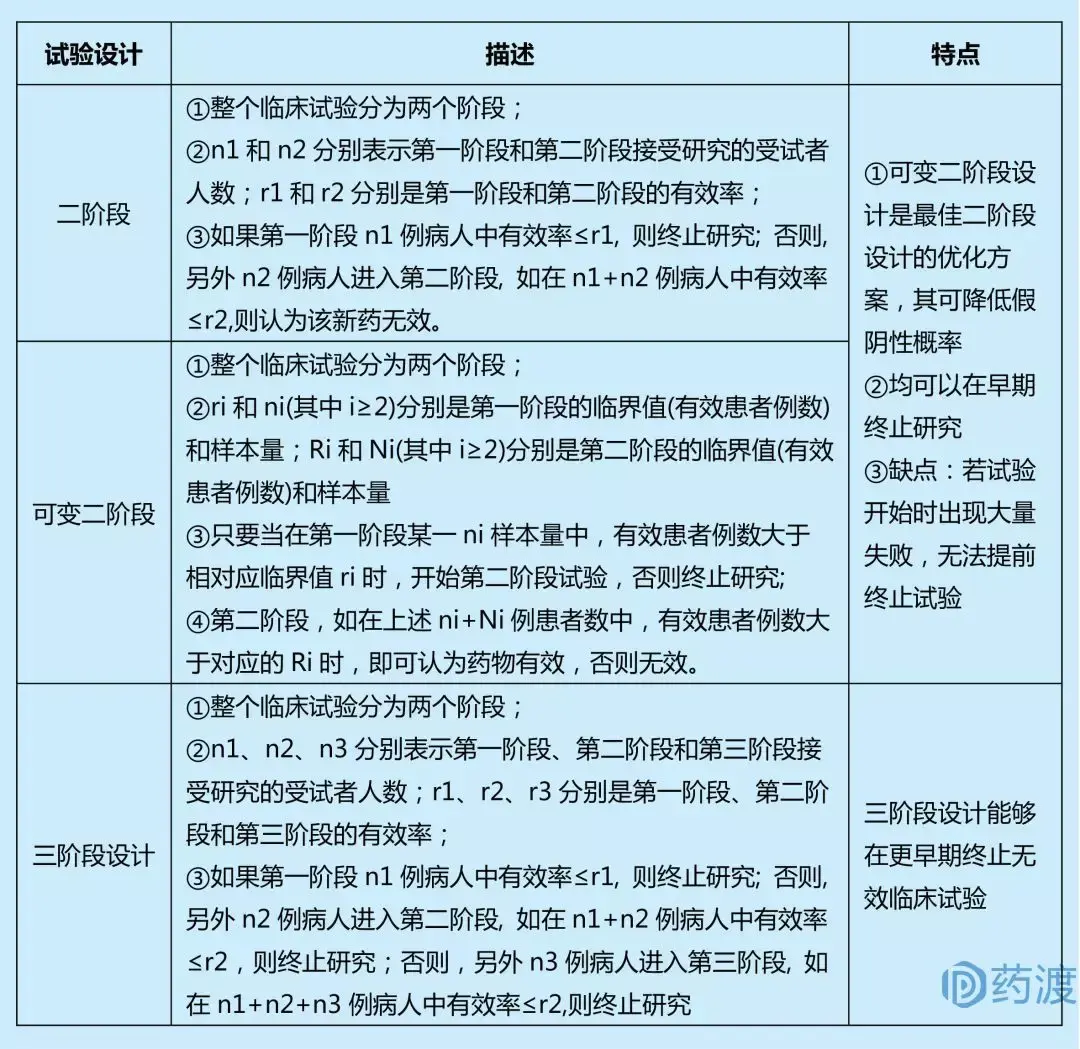
上述单臂多阶段试验计算具体样本量时,又分为最佳设计(optimal design)和极大极小设计(min-max design)。
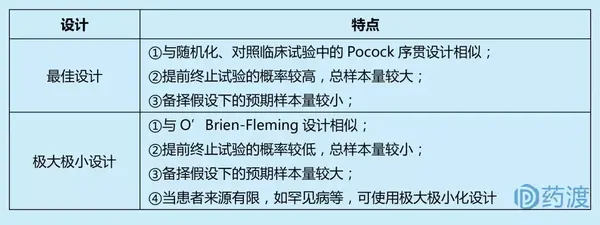
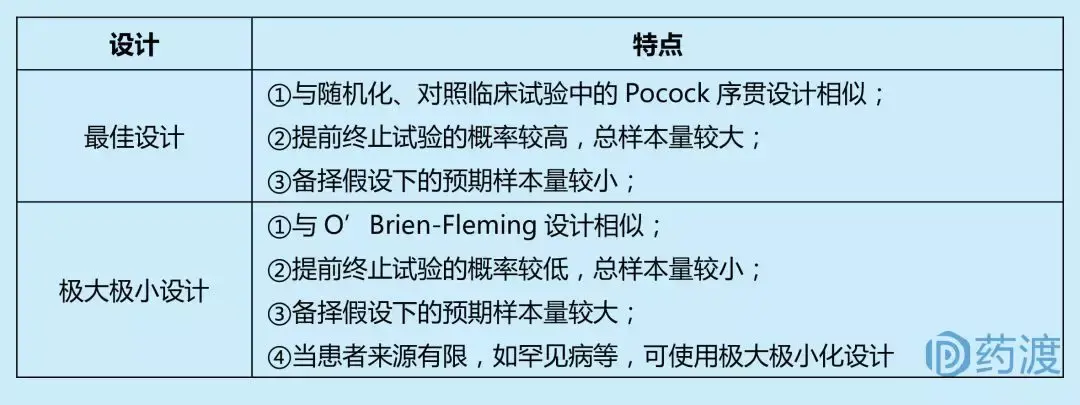
单臂多阶段设计一般用于探索性研究,其优点在于多阶段设计有明确的早期终止研究的准则,当试验药的有效率较低时,可以在早期终止研究,避免更多的受试者接受无效的治疗;多阶段设计也可用来早期淘汰不良反应高的药物。
3.2 随机对照II期临床设计
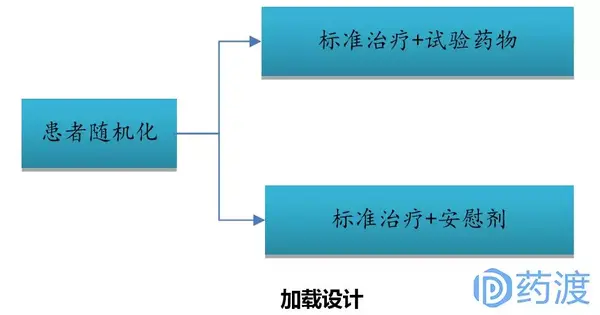
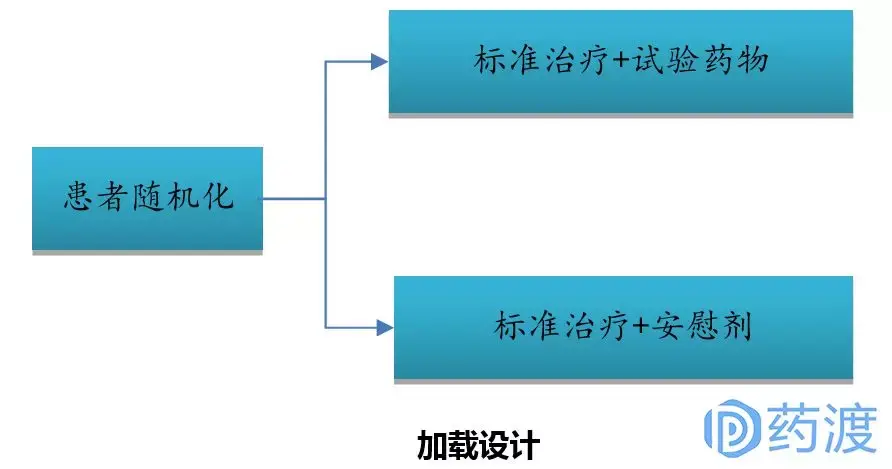
由于II期临床试验属于探索性人体试验,其样本量少,故大多数II期临床试验设计为单臂、非随机化,并且不设对照组,而是采用历史数据对照,这增加了对新药有效性判断的不确定性。为了降低III期临床试验失败的风险,作为前瞻哨所的II期临床鼓励采用随机对照设计,并且保证样本量具有一定统计学估算基础。尽管II期随机对照临床设计没有足够的统计把握度对新药和标准治疗间做出决定性的评价,但这种设计可以为有前景的新药优先进入III期试验提供量化依据。II期随机对照临床试验设计可应用于评价:多种剂量、多种给药方案、试验治疗和标准治疗对比的研究,为III期临床试验设计提供更加具有借鉴意义的数据。II期随机对照临床试验设计一般流程如下图所示。

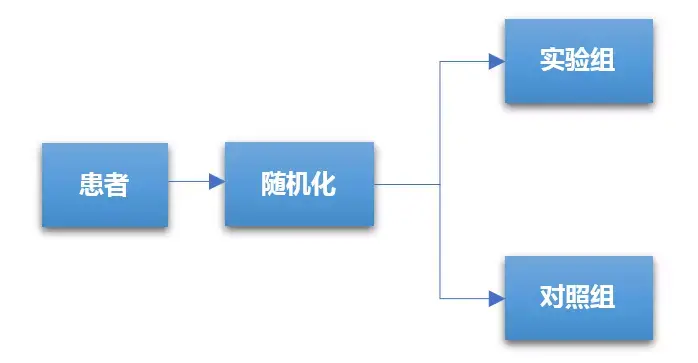
II期随机化对照临床试验主要目的是通过对所试验药物的有效率进行评估,从而选择有效率最佳的剂量、给药方案或候选药物进入III期临床试验。II期随机化临床试验所需的样本量不足以对试验药提供明确的优效性、非劣效性或等效性进行推断。
3.3 随机撤药设计
随机撤药研究是指通过接受一定时间受试药物治疗的对象出现疾病稳定性状态后,被随机分配继续使用受试药物治疗或使用安慰剂(即停用活性药物)治疗;继续接受药物治疗组和安慰剂组之间出现的任何差异都可以证明活性药物的疗效。随机撤药设计的优点是:病人经过安慰剂使用阶段比较短,伦理学风险被大大降低。随机撤药方法适合于复发性疾病发作的药物(例如抗抑郁药),抑制症状或体征(慢性疼痛、高血压、心绞痛)的药物临床对照试验等。
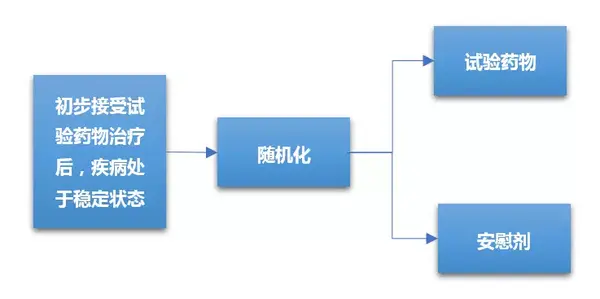
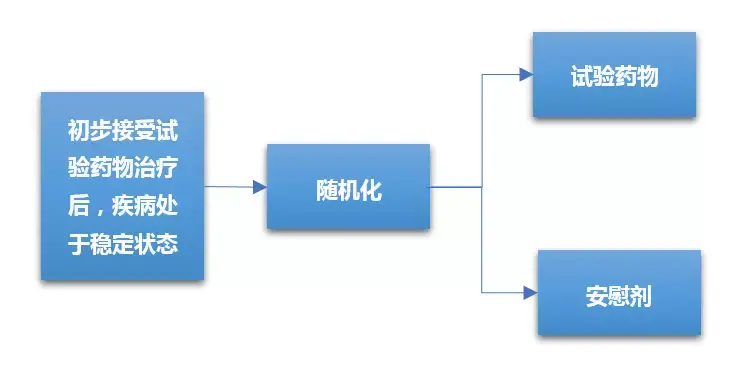
4 思考
II期临床试验一般是新药首次应用于目标患者,以观察新药的初步有效性、安全性数据。由于III期临床试验一般为大规模确证性试验,其常常为多中心、大样本、长周期的临床试验,是FDA评价、批准新药最关键的证据,而作为先头部队的II期临床试验对III期具有重大意义。合理科学的II期临床试验设计,不仅能够在早期辨析新药进一步开发的价值,同时还能够为III期临床试验推荐合理的临床定位、拟用于治疗适应症、适宜的纳入疾病人群的选择、主要疗效指标、安全性指标、给药剂量、给药方法、疗程等,可谓责任重大。迟发性毒性是II期临床试验中的问题之一,建议II期临床试验注意同时检测反应和毒性变量。另外,孤儿药或有重大突破性疗法新药在完成II期临床试验后,申办者可向FDA申请上市,FDA在评估患者利益/风险比后,可“有条件批准”该类新药基于临床II期数据提前上市销售;在新药上市销售期间,申报者仍需继续进行临床III期试验,以便产品最终获批。正所谓“秤砣虽小压千斤”,II期临床试验在整个新药临床试验中起到承上启下的作用。
参考文献:
1.临床试验的设计与分析
2.药物临床试验方法学
3.肿瘤新药Ⅱ期临床试验中的多阶段设计
4.抗肿瘤药物Ⅱ期临床研究设计考虑要点
5.中药新药临床研究一般原则
6.抗肿瘤药物的ii期临床试验--靶向治疗时代的常规设计和新策略介绍
新药临床试验设计路径:III期临床试验
1 序言
III期临床试验是新药临床研究阶段的关键性试验,是新药能否最终获批上市的临床基础。III期临床试验又称为确证性临床试验,其是为了进一步确证II期临床试验(探索性临床试验)所得到有关新药有效性和安全性的数据,为新药获得上市许可提供足够的证据。III期临床研究一般是关于更广泛人群、疾病的不同阶段,或合并用药的研究。另外,对于预计长期服用的药物,III期临床研究会进行药物延时暴露的试验。
2 III期临床试验全景
2.1 是什么(Who)
III期临床试验属于临床试验的治疗作用确证阶段,通过III期临床试验证明新药对目标适应症患者是安全有效的,其受益/风险比是可以接受的,为药物申报注册提供充分的依据,同时还为药品说明书和医生处方提供充分的数据。
2.2 为什么(Why)
II期临床试验受试者的样本量较少,其获得新药关于有效性和安全性的数据不足以支持新药获得上市批准。而III期临床试验可以通过足够多的受试样本量,进一步确证II期临床关于新药的疗效、长期安全性和受益/风险比,为新药最终获批上市提供确切的证据。
2.3 做什么(What)
III期临床主要用来回答一个问题:新药的受益/风险比如何?为回答该问题,III期临床试验一般是具有足够受试者样本量的随机盲法对照试验。III期临床试验也可以进行量-效关系的研究,同时也可以根据药物特点、目标患者的具体情况,进行药物相互作用等的研究。III期临床试验结束时需提供有统计学意义的结论,包括:新药目标适应症、所纳入的疾病人群、主要疗效指标、给药途径、用法用量及疗程、足够支持注册申请的安全性信息,并针对有效性安全性数据进行全面的风险/效益的评估等。另外,根据不同适应症或联合用药,申办者会将III期临床进一步细分为IIIa和IIIb期,申报者完成IIIa临床试验后即可申请上市批准,这样一般可以加快上市进度,提高市场收益;而通过IIIb临床试验可以进一步扩展新药适应症,加大市场收入。


2.4 怎么做(How)
试验原理: 一般通过新药与现有标准治疗的比较,III期临床试验分为优效性试验和非劣效性试验。试验过程常采用随机盲法、阳性对照试验;无市售阳性药物时,可选用安慰剂进行对照。
受试者: 目标适应症患者;
样本量: 一般为数百至数千人;
接下来主要介绍常见的III期临床试验设计。
3 III期临床试验设计
Ⅲ期临床试验一般采用随机、平行对照试验设计,确证新药在特定目标人群中的有效性和安全性。在具体临床试验设计方案中,试验设计类型的选择至关重要,因为这决定了样本量的估计、研究过程及其质量控制。因此,研究者应根据试验目的和试验条件的不同,选择不同试验设计方案。
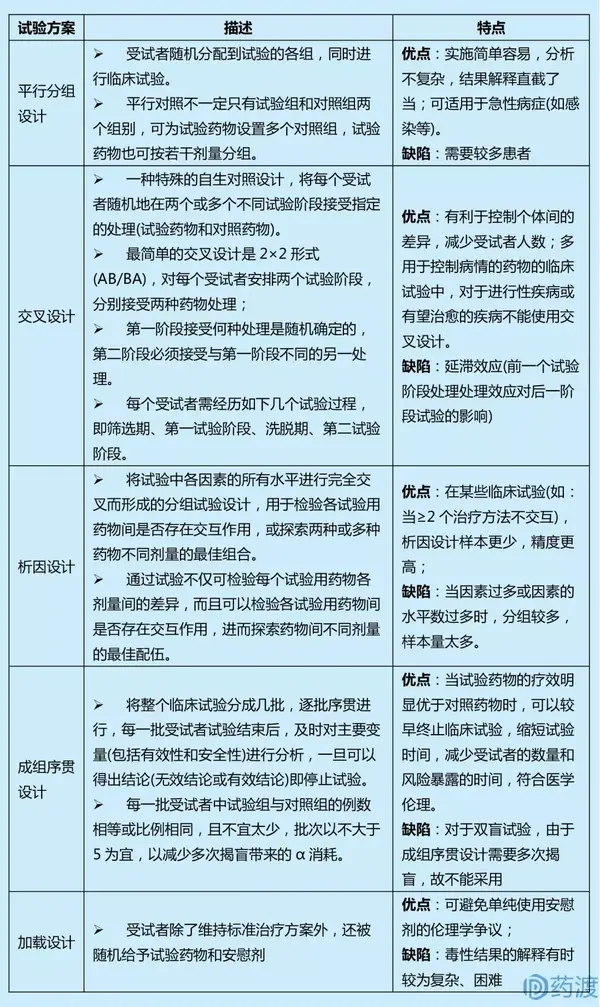
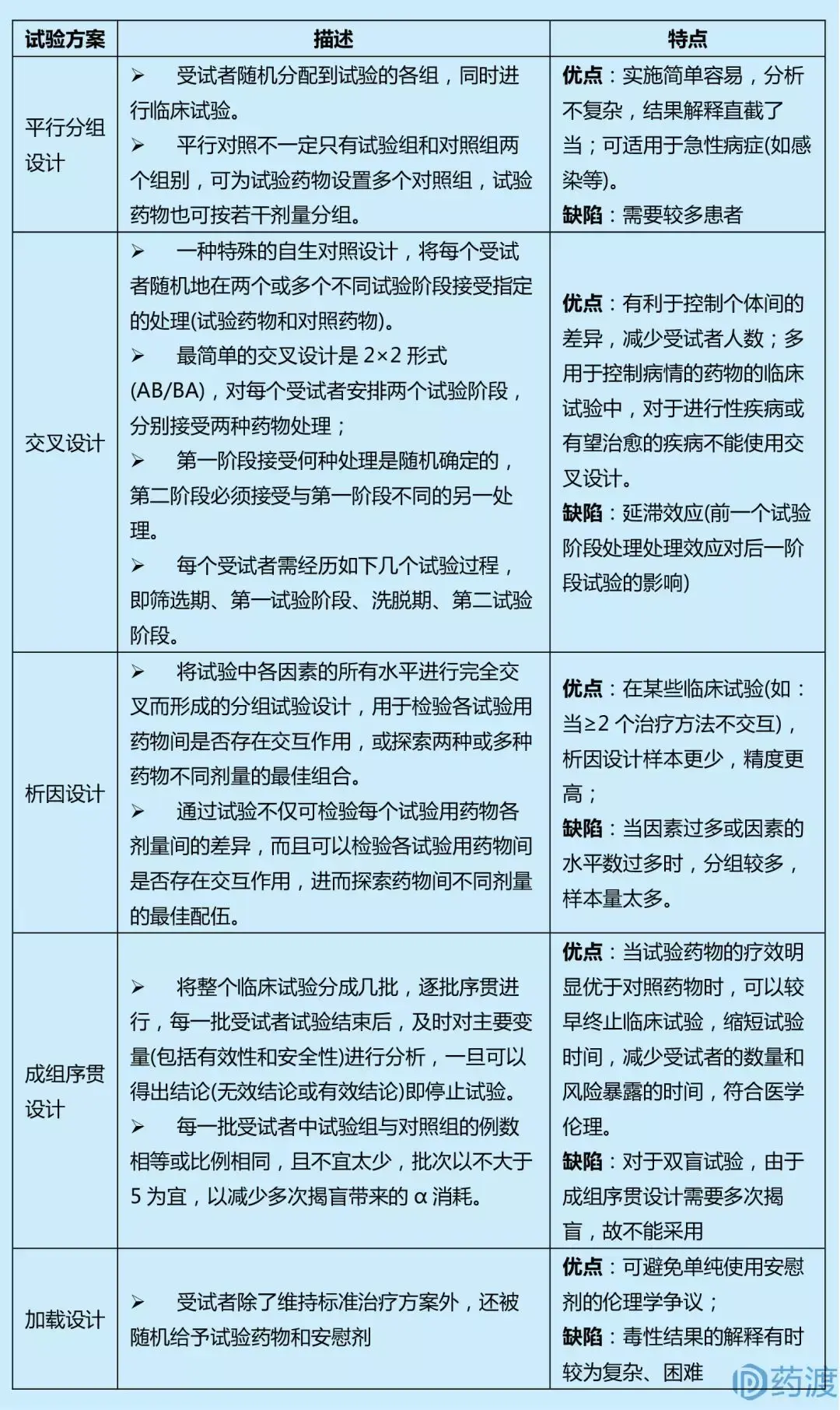

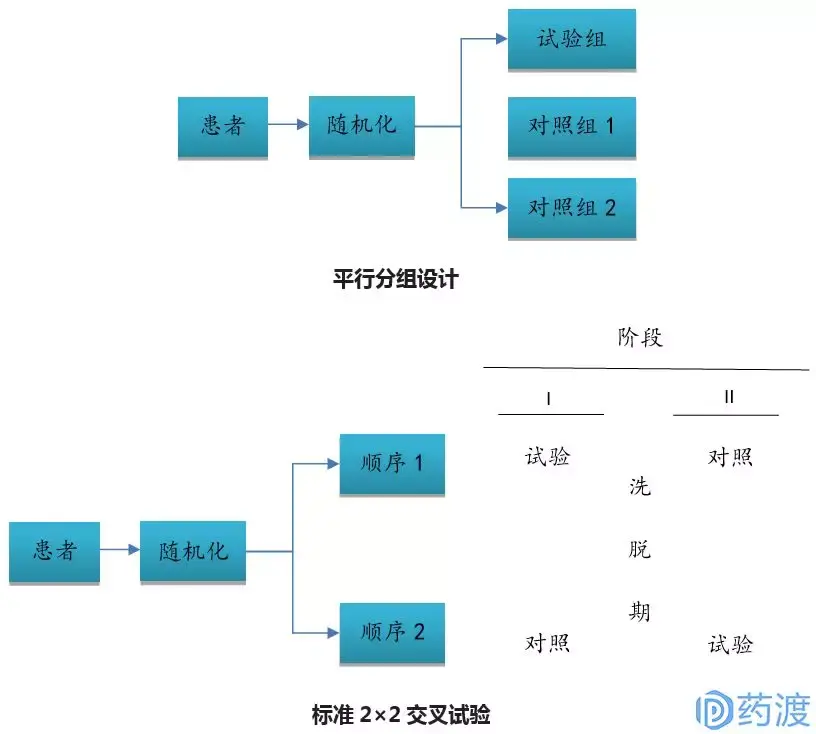
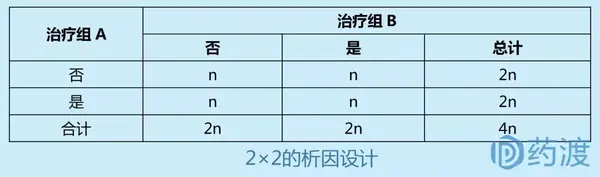
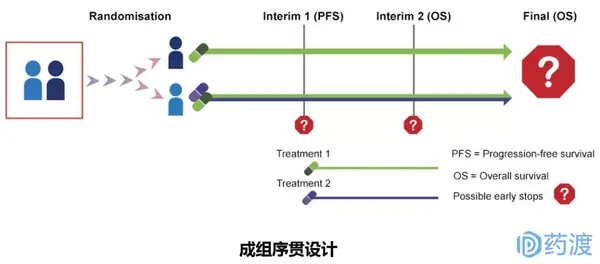
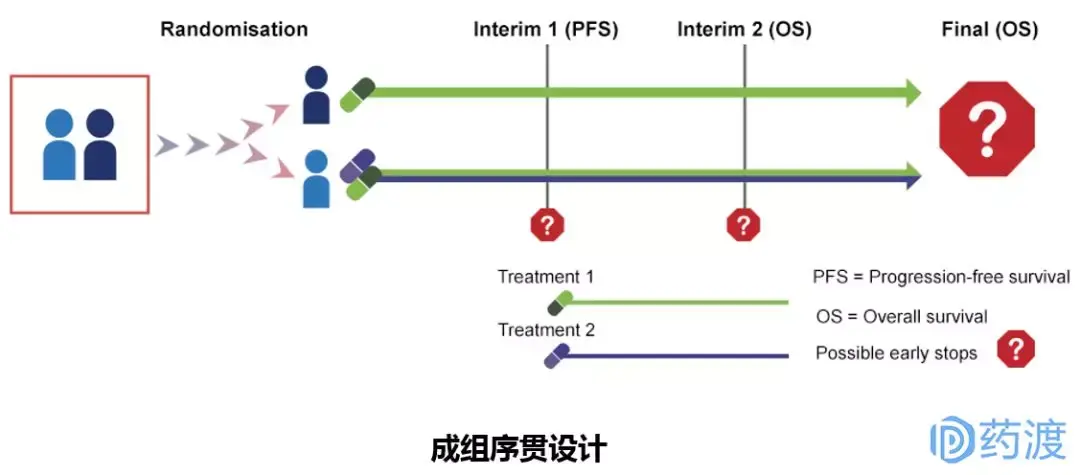
4 启示
一个新药经过首次数十人健康受试者的人体试验(First In Human,FIH),一直到5、6年后成千上百目标患者的III期临床试验,新药历尽磨难,可谓九死一生。整个过程历经人体耐受性试验、药代动力学试验、初步药效学试验、大规模确证性试验;其中可能还包括特定人群的考察(老人、儿童、功能缺陷等)、药物相互作用的考察等等。临床试验是新药是否批准上市的决定性阶段,任何一个环节出现问题,对于制药公司,尤其是初创公司打击是巨大甚至毁灭性的。同时,临床研究费用又是昂贵的,约占整个新药研发总费用的三分之二,高达数亿美金。正是由于临床研究的重要性和成本高昂,2007年FDA发布了《Target Product Profile — A Strategic Development Process Tool》,即基于目标产品特征(TPP)的产品开发策略。TPP是一种始于头脑的概念,即首先是申办者基于药物前期研究,通过设定未来产品的标签来定义新药开发目标,制定旨在支持产品标签(label)的具体临床试验;以TPP为主线和目标来全程、有效地指导和规划创新药临床开发和研究,提高申办者与管理当局在临床开发的各阶段(尤其是技术审评和会议)交流过程中效率,最终提升新药临床开发效率。
参考文献:
1、New approaches to clinical trials: Adaptive designs
2、美国抗肿瘤药物III期临床设计和评价介绍
3、Target Product Profile — A Strategic Development Process Tool
4、美国FDA “目标产品特性(TPP)指导原则(草案)”介绍
5、ICH E8
6、中药新药临床研究一般原则
7、药物临床试验方法学
8、临床试验的设计与分析
9、临床试验-方法学探究
10、药物临床试验的一般考虑指导原则
