

令人痛心疾首的PCR实验

不是所有的实验都有新手光环的,反正目前为止,我做rt-PCR是一次也没有做出来理想的效果。因为本实验室之前鲜有做PCR的,所以,大多时候,就是只能自己瞎琢磨,现在准备从头来学习。我尤其感谢我在医院检测科读研的我的好闺蜜,我每次一有啥问题就跑过去问他,他都不厌其烦地给我解释,虽然直至写这篇PCR实验总结的时候,我还没有完整地跑出一次好的结果,但是总算在每个方面都开始进步和有了收获,期待后面的不断调整可以让实验进展顺利把。
首先,做PCR第一步是RNA的提取,那要想得到准确的好的结果,提取好的质量的RNA保证RNA不会降解是实验成功的关键。
因此首先在提取RNA的过程中,就要严防RNA酶的污染。
关于RNase酶
是一些微小紧凑的蛋白,它们含有一些半胱氨酸残基能够形成许多分子内二硫键。因此在恢复至室温后,在无变性剂存在的条件下,变性的RNase会重新恢复其天然结构及部分功能。因此,RNase在 反复冻融 ,甚至 高压灭菌后 仍会保留相当的活性。这些酶类稳定的天然属性使它们对众多的去污染方法具有抵抗性,通常需要强力的化学方法来消除物体表面和溶液中的RNase。
避免RNA酶的污染的手段和途径可以见一下文章链接: 总结起来勤换手套,穿好实验服,避免在气流扰动条件下进行实验,无酶专用枪头和无酶专用枪。
那我们实验室目前提取RNA是买的试剂盒,进行RNA的提取。提取的RNA用微量核酸定量仪去进行浓度的测定,一般我每次RNA提取出来,浓度可能就是在150μg/mL左右,然后RNA的质量的话,初步可以用260/280以及260/230的比值来进行初步的判定。A260是核酸样品的最高吸收峰的波长,而A280是蛋白最高吸收峰的吸收波长,A230nm 是碳水化合物最高吸收峰的吸收波长。
A260/A280比值可进行核酸样品纯度评估:纯DNA的A260/A280比值为1.8,纯RNA为2.0。如果比值低,表示受到蛋白(芳香族)或酚类物质的污染,需要纯化样品。
A260/A230比值可进行核酸样品纯度评估:纯DNA和 RNA的A260/A230比值为2.5。若比值小于2.0标明样品被碳水化合物(糖类)、盐类或有机溶剂污染,需要纯化样品。
A260/A280和A260/A230
是核酸纯度的指示值。纯度好的DNA或RNA,在pH7-8.5 下A260 / A280的比值应该在2.0 或2.5。 纯净的样品比值大于1.8(DNA)或者2.0(RNA)。如果比值低于1.8 或者2.0,表示存在蛋白质或者酚类物质的影响。A230表示样品中存在一些污染物,如碳水化合物、盐(胍盐)等,较纯净的核酸A260/A230的比值 大于2.0 。
链接:
https://www.
jianshu.com/p/f91f88144
175
来源:简书
我之前每次提的RNA按照试剂盒的指示去设置程序,我发现260/230的值总是很小,可能是有炎或者有机溶剂的污染。后面我想来觉得最有可能的还是受到了有机溶剂的污染,因为我发现空管离心两分钟可能不足以把有机溶剂完全去除,后面我尝试空管离心四分钟,这个值多数大于2,虽然 还有几个样品出现了值小于2的情况,说明,很可能之前的按照说明说上的操作来做的话还是很有可能存在有机溶剂污染的情况。
提取了RNA开始做PCR了,还有一大堆问题。
比如一开始我做了好几次,重复了很多遍,PCR的融解曲线永远是这个样子的
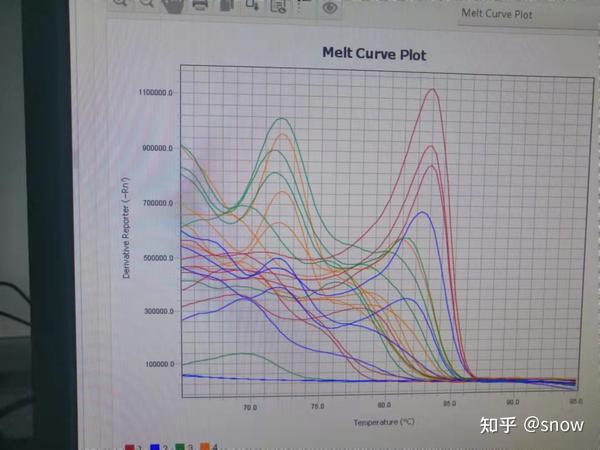

我一开始怀疑,是不是自己的RNA提的不好,发生降解了,或者自己没有在超净台做,导致实验结果乱七八糟,然后。。。我看到了有人说对于qRT-PCR来说,只要是样本不发生很离谱的降解,对于300bp这样微小的转录不会有太大影响的,出现这种可能的最大原因,就是引物不行。
引物不行??!!
Excuse me???
引物可是我找的参考文献找的,怎么会不行呢。
我闺蜜建议我自己设计引物,还有之前曾有为数不多一个实验室师姐跑过PCR,她的内参引物我可以拿过来跑一下看看,是不是真的是引物有问题。于是,通过B站教学以及我闺蜜的帮助,我学习了在NCBI上去设计我目标基因的引物,然后拿来我师姐之前跑过的内参引物,跑了重新跑了下,这次果然:
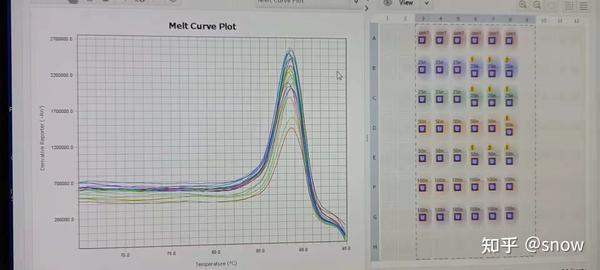
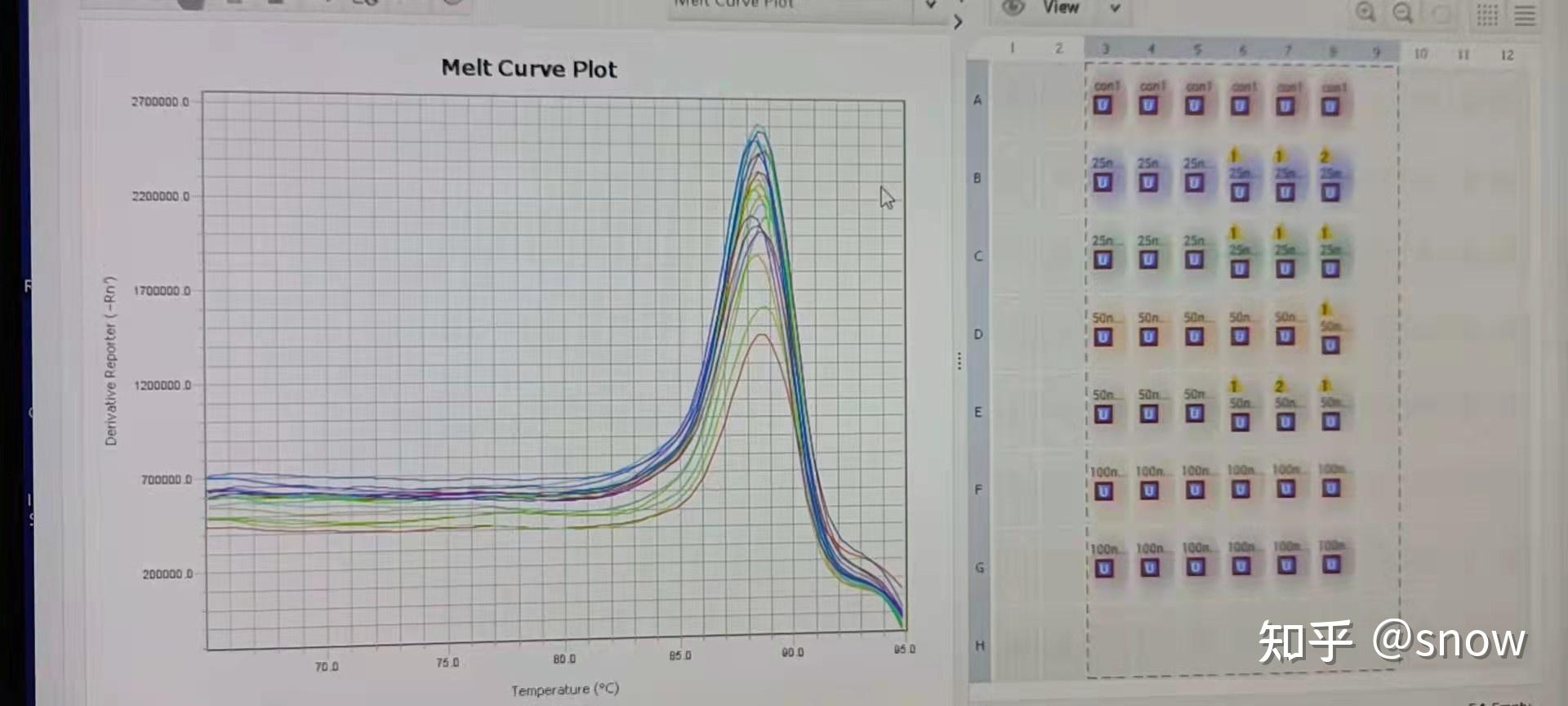
无论最终结果怎么样叭,至少融解曲线是正常了~
公司设计引物20元一对还不保证成功率,既然如此,不如自己花半个小时学习一下怎么用NCBI设计引物和blast引物,这样至少以后每次跑PCR都省了引物设计的钱,还可以完全被文献的参考引物所蒙蔽。随便是B站还是百度都有很多教你如何设计的教学资源,想学的小伙伴可以自己去找啦~
那设计好了引物,做qPCR我还是遇到了一大堆尚且没有解决的问题,呜呜呜。
比如内参和目标基因的CT值还是比较大的。
详细吐槽我的结果之前,还是需要了解一下实时荧光定量PCR的原理。
Ct值是PCR的最重要的表现形式,那么什么是Ct呢?
这里的C代表的是Cycle,而t代表的是threshold。他其实表示的是qPCR扩增过程中,扩增产物的荧光信号达到设定的荧光阈值时的所对应的扩增循环数(Cycle Threshold)。
做了几次PCR,参照文献上选的引物,就完全是跑不出来的,我一开始一直怀疑是自己操作是不是有问题,在一般的环境中进行操作,导致我的RNA降解了,后来,经常跑PCR的同学给我说,不要用文献里面的PCR引物,自己设计或者是找公司设计,十分简单。


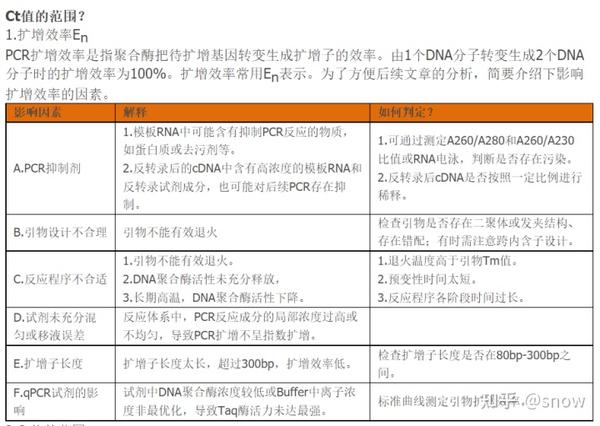

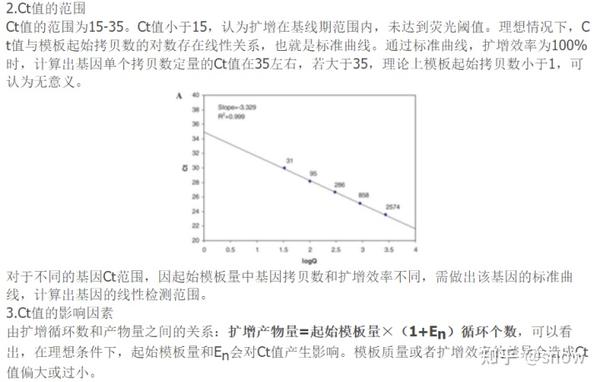
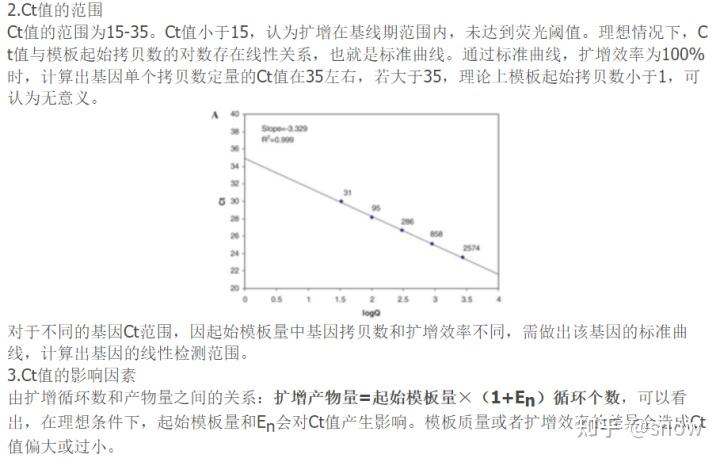

那么我前后做了好几次PCR的话,其实我每次做出来的结果都是一样的,就是Ct值过大,那么Ct值过大的话,有可能原因就是模板浓度太低,所以我又重新提高了模板的浓度,但是结果反而和预期相反了,那么感觉这就可以排除模板浓度低的原因,其实反而感觉更有可能是存在抑制物的原因,所以下次做PCR的时候,可以尝试将cDNA进行梯度稀释。
暂时的话,我先更新到这里,等明天看我重新梯度稀释后看结果了叭~
2021年9月22日重新更新
我今天又跑了一遍PCR(满怀希望地),因为感觉确实可能是存在抑制剂,梯度稀释的那种,结合我之前做的几次结果,相当于我做了稀释2,4,8以及16倍的样品,其实几次来说的话,感觉还是第一次做的相当于2倍稀释的效果比较好,后面稀释倍数太多了,虽然抑制剂的浓度减少了,但是相当于它的模板量也减少了。这就说明模板质量很重要,因为在模板质量存在问题的条件下,提高模板的数量和降低抑制剂的浓度是没有办法兼顾的。但是问题是我之前提的RNA,拿给别人做实验也没有存在这种问题,这样来看,提取的模板质量应该没有存在特别大的问题。
所以,,,,
天呐,为什么我做个PCR这么不顺利?
今天一个卖qPCR的销售来推荐产品,我现在打算去排除一下我之前买的试剂盒本身转录效率的原因,还有就是关于我的qPCR的程序虽然是按照参考书上面来的,是不是还可以优化呢???
还有我再试试提高一下体系的体积试试,再跑不出来,小夏得哭了。
-----------------------------------------------------------------------------------------------
2021年9月24日更新
昨天又跑了PCR,哈哈哈,这次终于跑的行了,至少内参和处理组的目标RNA的Ct值都在适宜定量范围之内了。
经过已经记不清多少次的失败,终于已经得到了自己比较满意的结果。
这次我也来复盘一下这次做实验终于做成功的过程,也给各位还在PCR实验上苦苦挣扎的朋友们提供一点自己的失败经验吧。
引物很重要!!!非常!!!
好的引物,跑出来的熔解曲线应该是漂亮的单峰,并且同一个目标基因对应的曲线的出峰位置应该差不多,如果熔解曲线出现双峰,很可能是形成了引物的二聚体或者是引物的特异性不好,出现了非特异性的扩增。所以早点换引物。
我用过两种品牌的试剂盒,都是要求要在冰上配置反应的体系的。
我之前想尽量提供干净的环境,避免模板RNA的降解,一直拿到我们细胞房的超净工作台上去操作,然后细胞房不允许带进去很大盆冰,我就用的是几块冰盒去保温的,但是感觉这样子效果不好,这也有可能是我之前扩增效率不太行的原因。后面我到实验室人少一点的试验台上去做的,用了大盆碎冰,充分冰浴,结果效果做出来才还不错。
所以说,这个冰浴的反应体系也很重要!!!
2021年10月20日更新
一个看上去周期不长的PCR实验,前前后后做了好多次了~
今天一个试剂公司来我们学校开了讲座,我又去听了,感觉收获还是挺多的,虽然大部分的原理都是之前已经自学过的,但是讲座中确实涉及到了很多很多我们实验中可能会遇到的问题,也相应的给出了一些解决方案,我觉得是非常有收获的,和大家一起分享一部分内容吧。
(其实工程师从实验原理到操作到结果分析都有涉及,甚至包括仪器知识比如ROX矫正的原理都有提到,但是为了不与上面的一些内容重复,我还是挑选一部分内容在这里展示叭)
实验分组

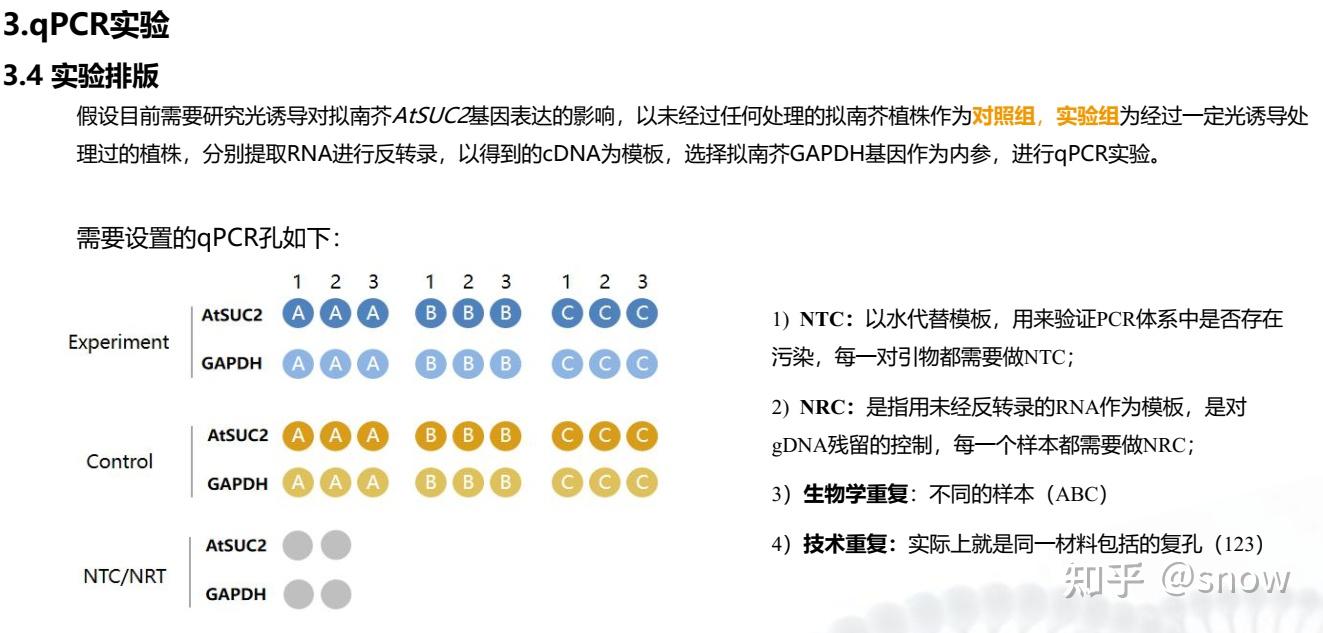
你一个比较标准科学的PCR实验需要包含到那些组别,说实话,这些分组里面有一些是我之前没有设置的,比如NTC以及NRT,这些组别的设置可以让你更好的排除你PCR过程中可能存在的一些问题。
数据分析
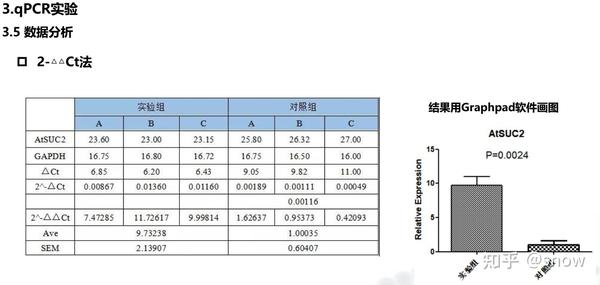

一般来说都是采用这种相对定量的分析方法,原理大家随便百度自己去学习一下叭。
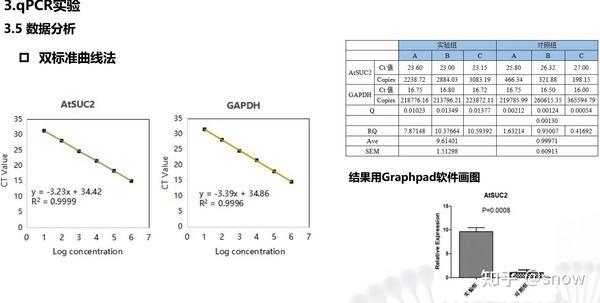

工程师解释到这种双标准曲线法一般来说是很少用到的,那么这种分析方法主要是适用于什么要的情况呢?主要是如果你的PCR结果感觉跑出来结果是不合理的,你可以看看是否存在这种情况,并用这种分析方法去进行数据处理看能否解决(这种方法主要是当内参基因与你的目标基因的扩增效率差别比较大的时候采用的)一般来说对于同样的扩增体系,这种扩增的效率是不会差别很大的。
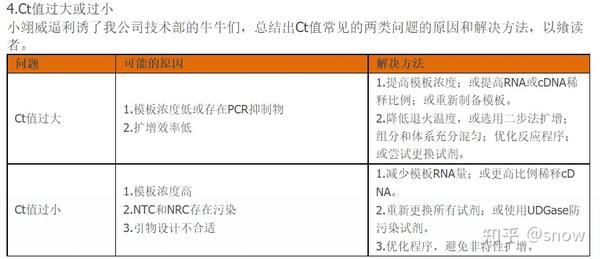
你有没有遇到过CT值偏大的情况?那可能是哪些原因?具体有哪些解决的办法呢?
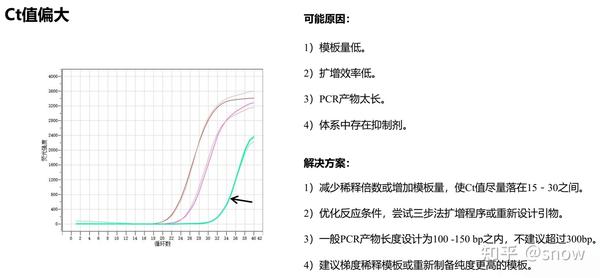

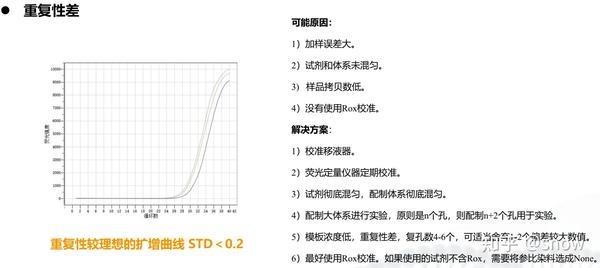

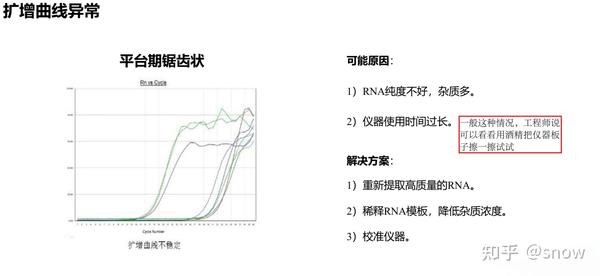

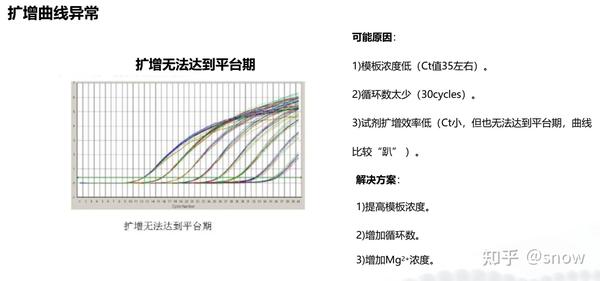

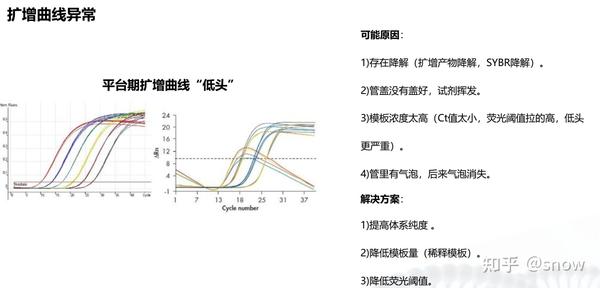

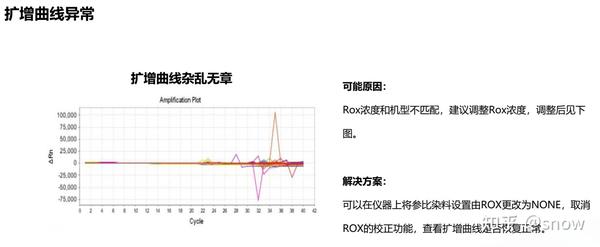

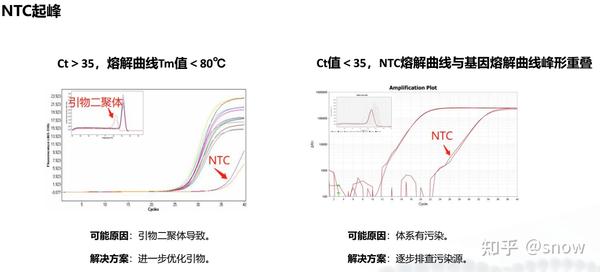
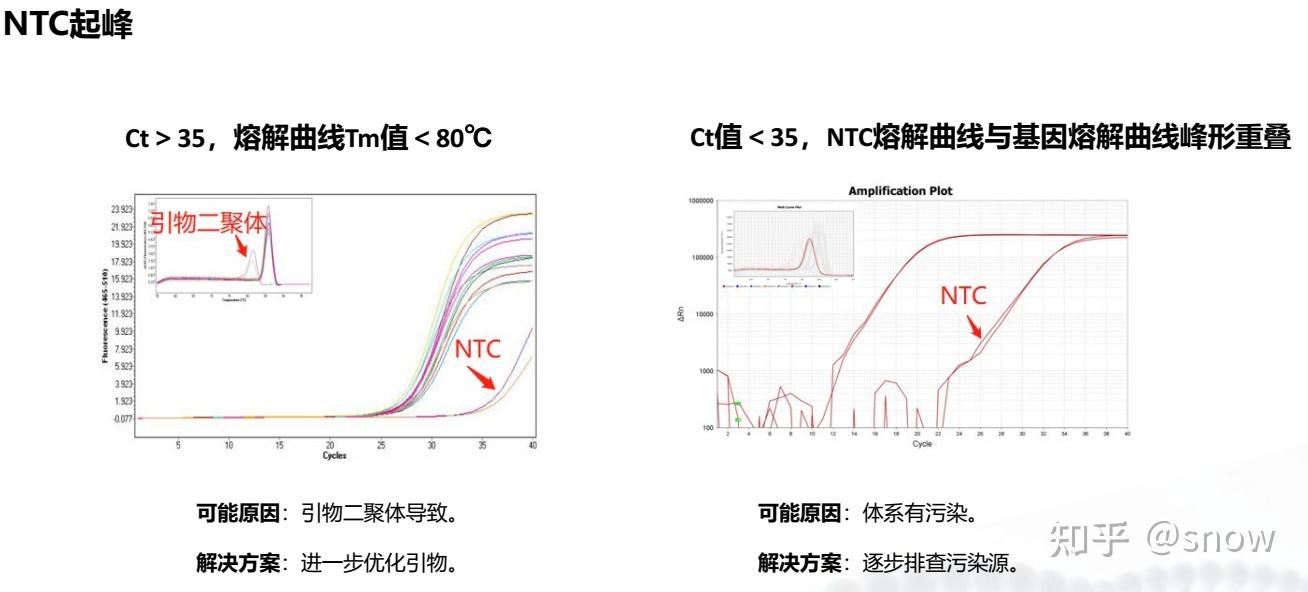




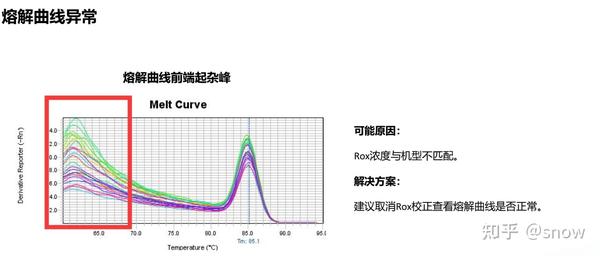
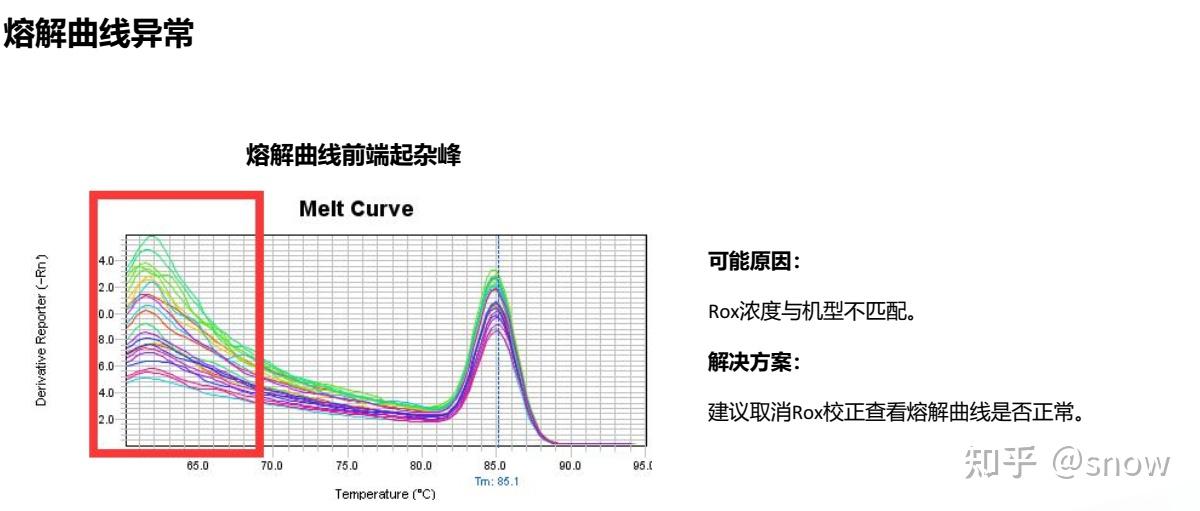
真的是你遇到没有遇到的情况都给你整理说明了,还提出了相应的解决方案,希望和我一样被PCR困扰的朋友们能从这里找到自救之路,大家一起加油叭。
那既然采用了人家的智慧,还是提一下这里智慧结晶的提供者(翌圣),不然我心理也过不去,反正用心做产品和售后的公司都值得尊重叭!