


Python版加权基因共表达网络WGCNA分析

在前面的教程中,我们介绍了使用omicverse完成基本的RNA-seq的分析流程,在本节教程中,我们将介绍如何使用omicverse完成加权基因共表达网络分析WGCNA。
环境的下载
在这里我们只需要安装
omicverse
环境即可,有两个方法:
-
一个是使用conda:
conda install omicverse -c conda-forge -
另一个是使用pip:
pip install omicverse -i https://pypi.tuna.tsinghua.edu.cn/simple/。-i的意思是指定清华镜像源,在国内可能会下载地快一些。
导入环境
import omicverse as ov
ov.utils.ov_plot_set()加权基因共表达网络分析(WGCNA)
加权基因共表达网络分析(WGCNA)是一种系统生物学方法,用于表征不同样品之间的基因关联模式,可用于鉴定高度协同的基因集,并基于基因集的内生性和基因集与表型之间的关联来鉴定候选生物标志物基因或治疗靶点。目前引用量已超过15,000。但Python中完成WGCNA分析相关的包仍是空白。我们根据WGCNA的原理,从底层上复现了原版WGCNA算法。
加载数据
在这里,我们选择WGCNA原版的演示数据来进行分析,数据可以在github上进行下载。
import pandas as pd
data=ov.utils.read_csv(filepath_or_buffer='https://raw.githubusercontent.com/Starlitnightly/ov/master/sample/LiverFemale3600.csv',
index_col=0)
data.head()
相关性矩阵计算
WGCNA的第一步是计算基因间的相关性矩阵,这里我们采用皮尔森系数的计算方法,来完成基因间的直接相关性矩阵计算。
gene_wgcna=ov.bulk.pyWGCNA(data,save_path='result')
gene_wgcna.calculate_correlation_direct(method='pearson',save=False)
在 pyWGCNA 模块中,我们需要将
直接相关矩阵
转换为
间接相关矩阵
来计算软阈值,软阈值可以帮助我们将原来的相关网络转换为无尺度网络
gene_wgcna.calculate_correlation_indirect(save=False)
gene_wgcna.calculate_soft_threshold(save=False)
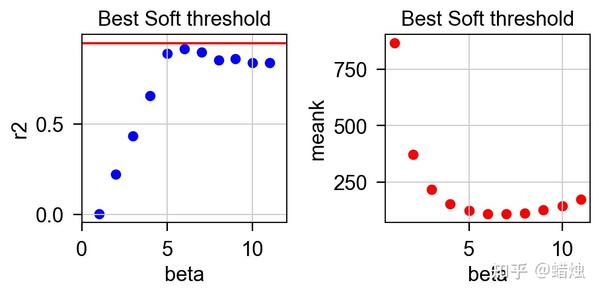
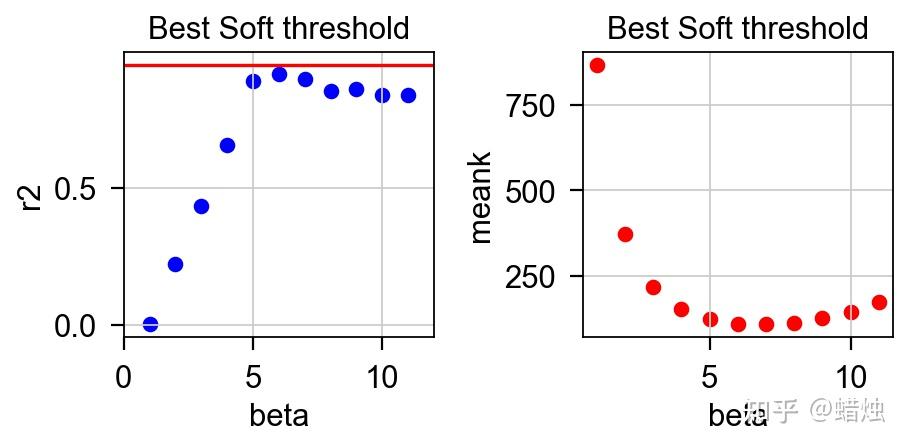
左边的垂直坐标是无尺度网络的评估指标
r^2
。R2越接近1,网络就越接近无尺度网络,通常需要
r^2
> 0.8或0.9。右侧垂直坐标为平均连通度,随 β 值的增加而减小。将这两个图结合起来,通常选择
r^2
首次达到0.8或0.9或更高时的 β 值。利用 β 值,我们可以根据方程将相关矩阵转换成邻接矩阵。
然后我们构造拓扑重叠矩阵。
gene_wgcna.calculate_corr_matrix()
共表达网络分析
在获得基因间的拓扑重叠矩阵后,我们使用动态剪切树的方式来寻找基因间的模块。在这里,我们使用WGCNA作者发表的DynamicTree的算法来实现此功能。
gene_wgcna.calculate_distance()
gene_wgcna.calculate_geneTree()
gene_wgcna.calculate_dynamicMods()
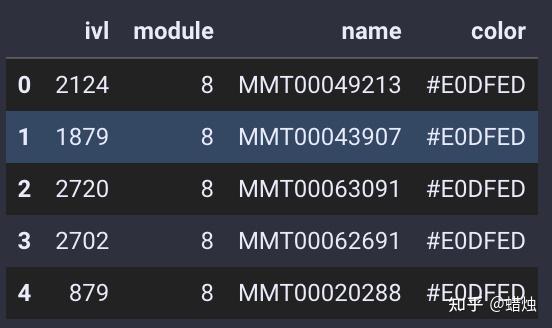
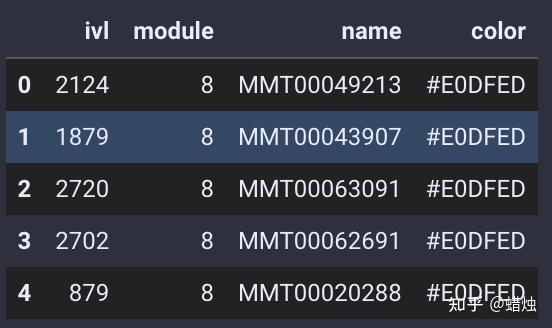
module=gene_wgcna.calculate_gene_module()
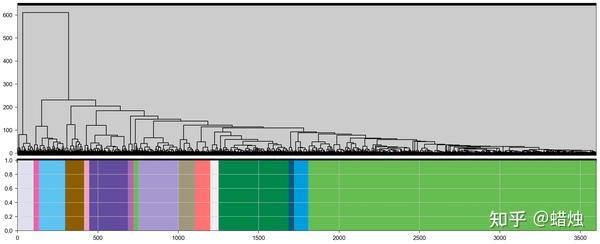
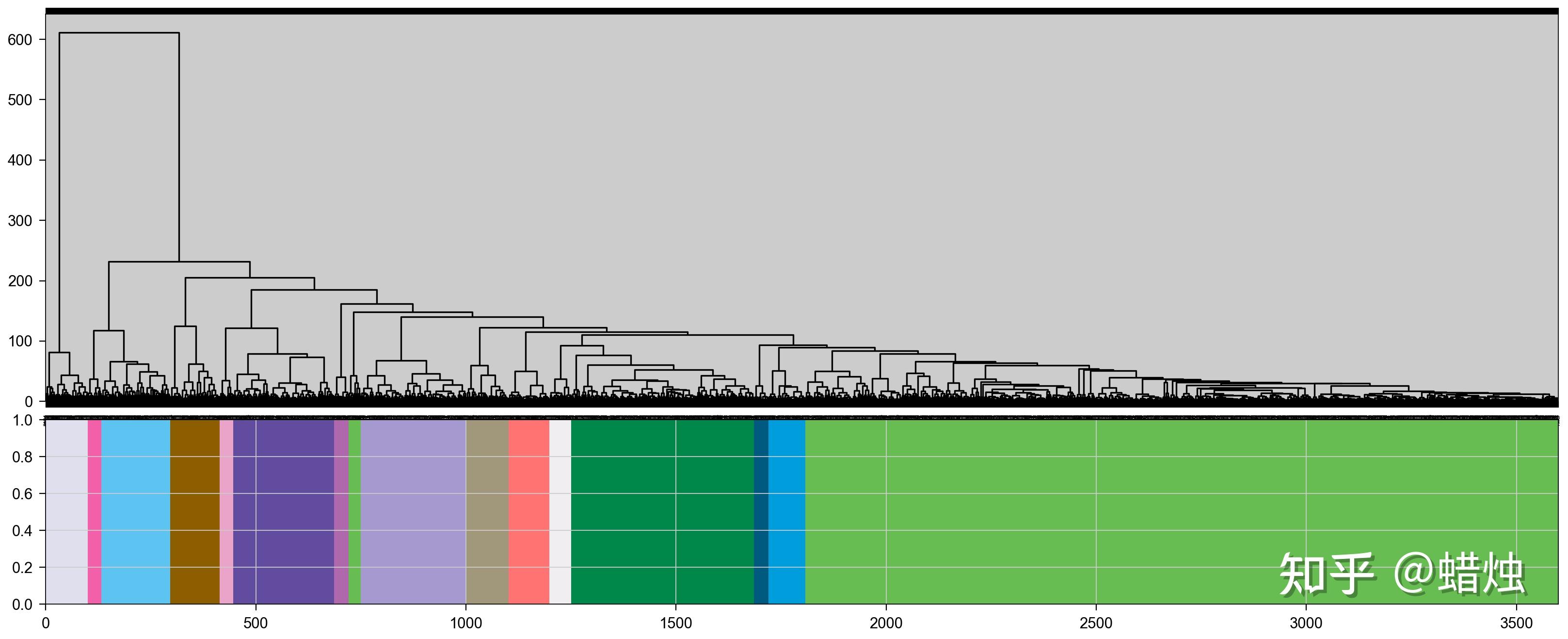
在这里,我们成功地计算了每个基因的模块,共有15个模块,它们的颜色都显示在图中。我们使用
.head()
查看每个基因所属的模块。
module.head()
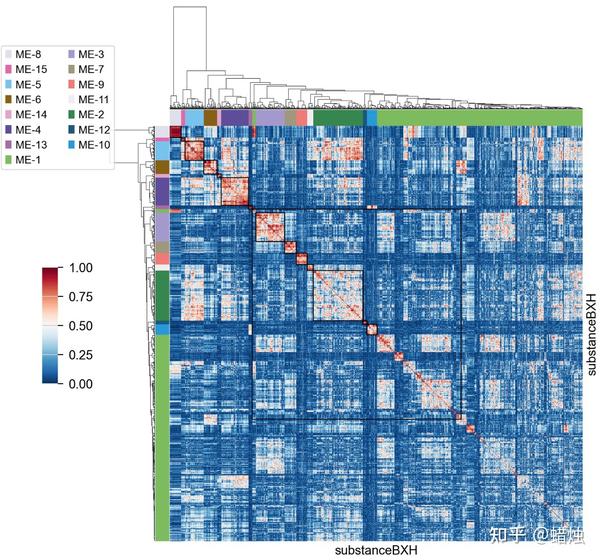
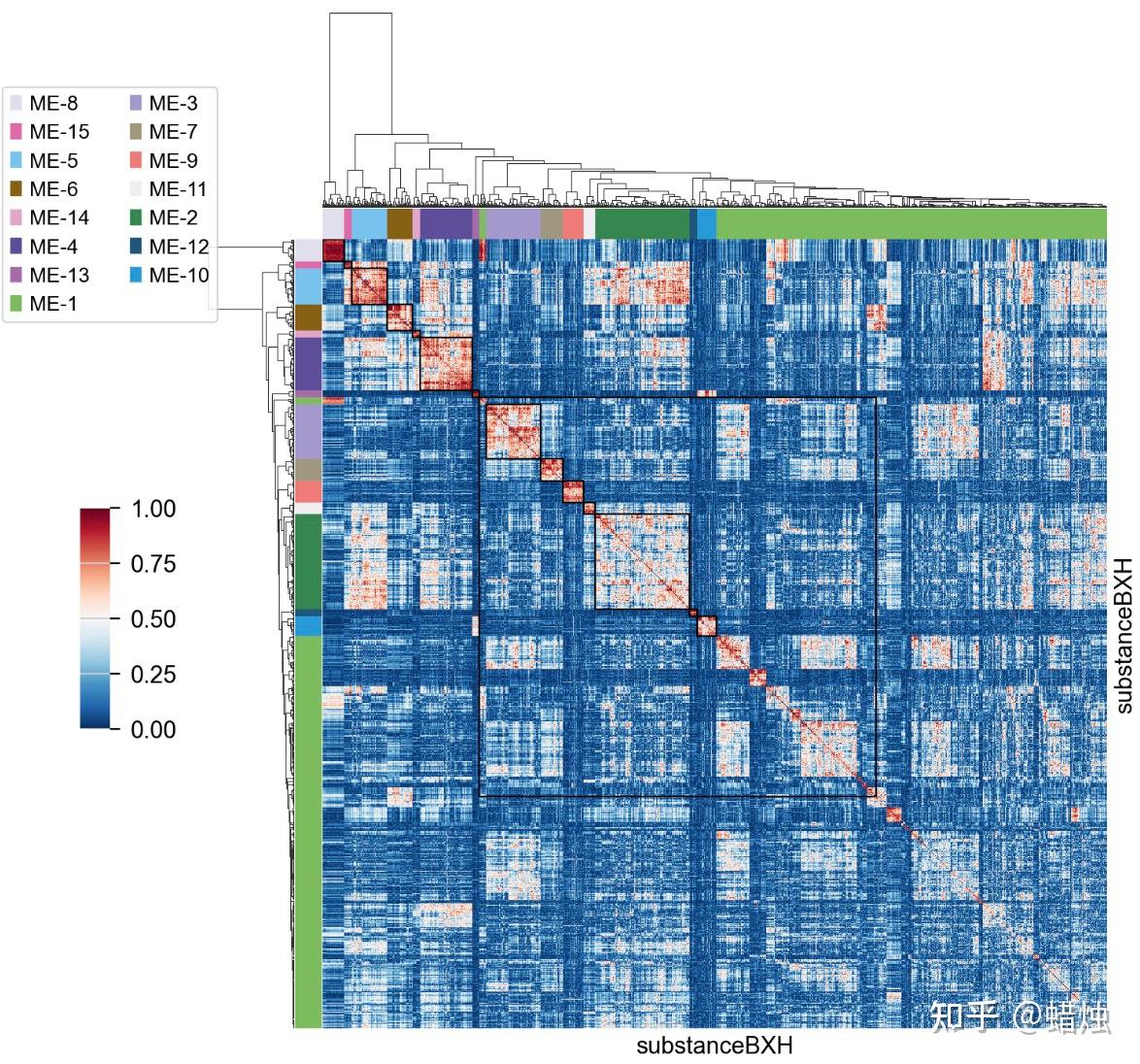
我们还可以使用
.plot_matrix()
来可视化拓扑重叠矩阵与模块之间的关系。
gene_wgcna.plot_matrix()
子模块分析
有时候我们对一个基因或一个通路的模块感兴趣,我们需要提取基因的子模块进行分析和定位。例如,我们选择了两个模块6和12作为分析的子模块
gene_wgcna.get_sub_module([6,12]).shape