


论文笔记5|AttentionDTA Drug–Target Binding Affinity Prediction by Sequence-Based Deep Learning ......


AttentionDTA: 基于序列的深度学习及注意力机制的药物-靶标结合亲和力预测
摘要
药物-靶标相互作用(DTRs)的识别对药物开发至关重要。这些方法的主要缺点是缺乏可靠的药物-靶标相互作用(DTAs)的二元分类问题的大量可靠数据。随着近年来DTRs和DTAs高亲和力结合数据的不断增加,DTAs识别可以被视为回归问题,这增加了公开的药物-蛋白质结合亲和力数据。这些方法往往比使用药物-靶标亲和力数据的方法提供更多的信息。尽管这些方法在结合亲和力预测中相当有效,但由于深度学习黑箱模型的缺点,这些模型在生物学上通常难以解释。在这项研究中,我们提出了一个基于深度学习的模型,名为AttentionDTA,该模型使用注意力机制来预测DTAs。与使用3D结构药物-靶标复合物或图表示药物和蛋白质的模型不同,我们工作的新颖之处在于使用注意力机制关注在预测其亲和力时重要的药物和蛋白质序列子序列。我们使用两个独立的一维卷积神经网络(1D-CNNs)来提取药物的SMILES字符串和蛋白质氨基酸序列的语义信息。此外,我们还开发并嵌入了一个双向多头注意力机制到我们的模型中,以探索药物特征和蛋白质特征之间的关系。我们在三个公认的DTA基准测试数据集Davis, Metz和KIBA上评估了我们的模型。结果显示,注意力机制模型在不同的评估指标下胜过了现有的最先进的深度学习方法。值得注意的是,我们在IC50数据集上测试了我们的模型,该数据集提供了药物和蛋白质之间的结合位点信息,以评估我们的模型定位结合位点的能力。最后,我们可视化了注意力权重以展示模型的生物学意义。AttentionDTA的源代码可以从以下网址下载: https://github.com/zhaoqichang/AttentionDTA_TCBB 。
1 引言
新发现的药物-靶标关系(DTRs)在药物发现和开发中至关重要。目前,许多化合物尚未作为药物使用。据统计,DrugBank数据库中大约有13,580个药物条目,其中包括2,635个批准的小分子药物,1,378个批准的生物制剂和6,373个实验性药物。在大约20,000个人类蛋白质中,只有3,150个与这些药物有关,而人类药物靶标的估计数量大约为4,500个,约占人类基因组的22%。因此,需要验证大量潜在的DTRs。检测化合物与疾病相关蛋白质之间未知关系对于药物开发和药物重新定位至关重要。【1】【2】【3】【4】
DTR预测的一个常用方法是体外筛选实验,这些实验成本高,劳动强度大,并且失败率高。此外,考虑到庞大的化学和蛋白质组空间,验证每一对可能的药物-靶标组合不是一个现实的方法。可以通过引入虚拟筛选(VS)来加速这一过程。因此,通过计算方法预测DTRs引起了越来越多的关注。据报道,基于深度学习的方法在DTR预测方面比传统的基于机器学习的方法更为强大。这是因为它能够通过分层学习数据中的复杂和非线性特征来提取隐藏信息。随着近年来大量生物活性数据的发布,基于深度学习的方法使得可以在大规模上预测药物与靶标蛋白之间的关系成为可能。
大多数基于深度学习的方法将药物-靶标相互作用(DTIs)的预测视为一个二元分类问题,即确定一对药物-靶标是否相互作用。不同的深度学习模型已被开发来预测DTIs并取得了良好的性能,包括支持向量机[13] [14],深度神经网络[15] [16],堆叠自编码器[17] [18],和深度信念网络[19] [20]。Du等人[21]提出了一个框架,在该框架中宽模型是一种泛化的线性模型,深模型是一种特征转换网络以及深度模型的集成。Zheng等人[22]提出了一个名为DTI-RCNN的混合神经网络,将循环神经网络(RNN)与卷积神经网络(CNN)结合。Lee等人[23]提出了一种称为DeepConv-DTI的方法,使用化合物的指纹字符串作为输入,采用不同大小的卷积核来捕获蛋白质的局部残基构象模式,有效降低了不当的卷积核尺寸风险。Tsubaki等人[24]开发了一种新的DTI预测方法,通过结合图形神经网络(GNN)来处理药物,CNN来处理蛋白质,并将注意力机制[25]整合到表示中。Cheng等人[26]提出了一个端到端的深度学习方法来预测基于图形表示的DTIs的多头自注意力机制。Rifaioglu等人[27]提出了一个大规模DTI预测系统,名为DEEPScreen。DEEPScreen采用化合物图像作为输入,并构建了一个基于卷积神经网络的药物图像分类子网,参考了经典的图像分类网络,以准确地预测小分子配体对单一靶蛋白的亲和力。研究人员还使用了其他方法来处理DTI预测。Zhang等人[28]提出了一个基于问答(QA)模型来预测DTIs,通过表示蛋白质的2D距离图(Image)和药物的分子线性符号(String)来预测DTIs。Inspirer和Transformer's [29]的研究能够捕捉两个序列之间的特征。Chen等人[30]提出了一种名为TransformerCPI的方法来预测DTIs。TransformerCPI使用了改进的卷积网络来代替原始的自注意力编码器,以更好地处理药物和蛋白质序列中的跨序列特征。
DTI预测中一个不可避免的问题是缺乏可靠的负例(即确定的非相互作用)【32】。从未标记的DT对集合中随机选择负例并不能保证模型能够很好地区分正例和未知样本。由于使用了不恰当的数据集,许多DTI预测方法被指出是根据配体模式而非药物-靶标相互作用特征来做出预测的,导致理论建模与实际应用之间的不匹配【30】。此外,在现实中,DTR是通过连续的尺度实验测量的,该尺度量化了药物与靶标结合的强度。生物化学相互作用源自于药物原子和受体蛋白质上氨基酸的相互作用。结合亲和力受到非共价分子相互作用的影响,如范德华力、疏水作用、氢键和静电相互作用。药物-靶标相互作用预测的优势在于避免了负样本选择偏差的影响和数据集中隐藏的配体偏见,并提供了更实用和有用的信息【9】【32】。近年来,深度学习在直接从原子结构预测药物相互作用方面取得了良好进展。AtomNet【33】是第一个处理3D结构的深度学习模型,而不是使用传统的二维卷积神经网络(3D-CNNs)。Gomes等人【34】提出了一种自适应卷积神经网络(ACNN)来预测结合亲和力。ACNN包含了空间金字塔池化,用于编码局部化学环境和提取与生物分子识别相关的特征。受SqueezeNet【35】启发,Jimenez等人【36】提出了一个端到端框架,基于3D-CNNs预测药物-靶标绝对亲和力。为了减少计算复杂性和提高预测性能,Li等人【37】提出了一种基于3D-CNNs的有效且轻量级的神经网络,自动提取与药物-靶标相互作用相关的原子模式。然而,这些方法需要3D结构数据,而大多数药物-靶标对并非源自实验解决的晶体结构。此外,3D网格表示的药物-靶标复合物是稀疏的且计算成本高。大规模使用3D基于序列的方法可以有效地避免3D结构的缺失和计算成本问题。DeepDTA【38】是一个基于图形的序列方法,使用药物SMILES字符串和氨基酸序列作为输入,包含两个独立的1D-CNN模块来有效预测DTAs。WideDTA【39】利用药物特征、配体最大公共子结构(LMCS)和蛋白质基序和结构域提高DeepDTA的性能。GraphDTA【40】采用药物的图形结构作为输入,使用卷积图神经网络(GCNs)、图注意力网络(GAT)【41】和图同构网络(GIN)【42】来捕获相关特征。尽管这些深度学习方法表现出了卓越的预测性能,但它们的黑盒特性限制了它们在生物学角度上的可解释性。
为了解决这个问题,我们提出了一个端到端的模型,名为AttentionDTA,来预测药物-蛋白质对的结合亲和力。本文扩展了之前发表的一篇会议论文[44]。之前的会议论文提出了两种注意力计算模式,即共注意力和交叉注意力,并在DeepDTA的设置下进行了比较实验S1。在本文中,我们扩展了这项工作。首先,我们开发了一个新颖的双侧多头注意力机制,并设计了消融研究来探索注意力计算模式对模型性能的影响。其次,我们重新设计了实验比较程序。
我们提出的模型AttentionDTA在三个基准数据集上执行,即Davis [45]、Metz [46]和KIBA [47]数据集,并与两种最先进的方法,即DeepDTA和GraphDTA进行比较。我们设置了四种不同的数据分割设置,以进行全面比较。第三,为了评估我们模型定位结合位点的能力,我们在IC50数据集[48]上测试了我们的模型,该数据集提供了药物和蛋白质之间的结合位点。此外,为了更好地显示可视化效果,我们通过将注意力权重映射到蛋白质序列上,添加了新的可视化方法。
2 材料和方法
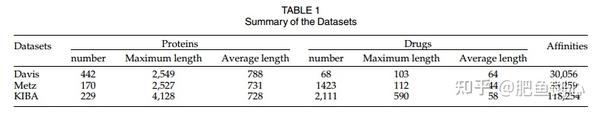
2.1 数据集



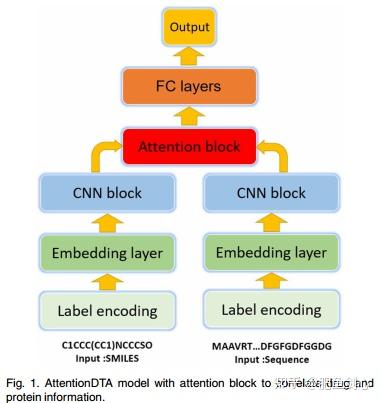
2.2 AttentionDTA模型
AttentionDTA的设计灵感来自于不同药物在同一蛋白质上有不同的结合位点,反之亦然的概念。如前所述,我们采用了一种流行的深度学习架构,1D-CNN,来从序列中提取有用信息。1D-CNN能够捕获整个空间中重要的局部模式。通过在蛋白质或SMILES字符串上滑动卷积滤波器,氨基酸的不同组合或药物的亚结构被捕获。为了模拟原子和氨基酸之间的非共价相互作用,我们在我们的模型中整合了一个双侧多头注意力机制。我们在这个模块中计算药物和蛋白质子序列之间的注意力分数。子序列的向量表示根据注意力分数被强化或削弱。
注意力机制通过允许模型动态地关注药物和靶标中的某些部分,这些部分有助于有效预测DTAs,引入了相关性的概念,但不可避免地,这增加了计算资源的额外消耗。在此之后,捕获蛋白质相关信息的药物表示和捕获药物相关信息的蛋白质表示被获得。AttentionDTA模型的工作流程如图1所示。
# 这段代码定义了一个名为 `AttentionDTA` 的神经网络模块,它是为药物-靶标亲和力预测(Drug-Target Affinity,DTA)设计的。这个模块使用了多头注意力机制和卷积神经网络(CNN)来处理药物和蛋白质的序列数据。这个模型结合了嵌入层、卷积层、注意力机制和全连接层,用于处理和分析药物与蛋白质之间的相互作用。通过这个架构,可以有效地提取药物和蛋白质序列的特征,并预测它们之间的亲和力。下面是对代码的详细注释:
class AttentionDTA(nn.Module):
def __init__(self, protein_MAX_LENGH=1200, protein_kernel=[4, 8, 12],
drug_MAX_LENGH=100, drug_kernel=[4, 6, 8],
conv=32, char_dim=128, head_num=8, dropout_rate=0.1):
初始化AttentionDTA模块
:param protein_MAX_LENGH: 蛋白质序列的最大长度
:param protein_kernel: 蛋白质卷积核尺寸
:param drug_MAX_LENGH: 药物序列的最大长度
:param drug_kernel: 药物卷积核尺寸
:param conv: 卷积层的输出通道数
:param char_dim: 字符嵌入的维度
:param head_num: 注意力头的数量
:param dropout_rate: Dropout层的概率
super(AttentionDTA, self).__init__()
# 初始化各种参数
self.dim = char_dim
self.conv = conv
self.dropout_rate = dropout_rate
self.head_num = head_num
self.drug_MAX_LENGH = drug_MAX_LENGH
self.drug_kernel = drug_kernel
self.protein_MAX_LENGH = protein_MAX_LENGH
self.protein_kernel = protein_kernel
# 嵌入层,用于将字符编码转换为固定大小的向量
self.protein_embed = nn.Embedding(26, self.dim, padding_idx=0)
self.drug_embed = nn.Embedding(65, self.dim, padding_idx=0)
# 药物的卷积神经网络层
self.Drug_CNNs = nn.Sequential(
nn.Conv1d(in_channels=self.dim, out_channels=self.conv, kernel_size=self.drug_kernel[0]),
nn.ReLU(),
nn.Conv1d(in_channels=self.conv, out_channels=self.conv * 2, kernel_size=self.drug_kernel[1]),
nn.ReLU(),
nn.Conv1d(in_channels=self.conv * 2, out_channels=self.conv * 3, kernel_size=self.drug_kernel[2]),
nn.ReLU(),
self.Drug_max_pool = nn.MaxPool1d(self.drug_MAX_LENGH - self.drug_kernel[0] - self.drug_kernel[1] - self.drug_kernel[2] + 3)
# 蛋白质的卷积神经网络层
self.Protein_CNNs = nn.Sequential(
nn.Conv1d(in_channels=self.dim, out_channels=self.conv, kernel_size=self.protein_kernel[0]),
nn.ReLU(),
nn.Conv1d(in_channels=self.conv, out_channels=self.conv * 2, kernel_size=self.protein_kernel[1]),
nn.ReLU(),
nn.Conv1d(in_channels=self.conv * 2, out_channels=self.conv * 3, kernel_size=self.protein_kernel[2]),
nn.ReLU(),
self.Protein_max_pool = nn.MaxPool1d(self.protein_MAX_LENGH - self.protein_kernel[0] - self.protein_kernel[1] - self.protein_kernel[2] + 3)
# 多头注意力机制层
self.attention = mutil_head_attention(head=self.head_num, conv=self.conv)
# 全连接层及激活层
self.dropout = nn.Dropout(self.dropout_rate)
self.relu = nn.ReLU()
self.tanh = nn.Tanh()
self.leaky_relu = nn.LeakyReLU()
self.fc1 = nn.Linear(192, 1024)
self.dropout1 = nn.Dropout(self.dropout_rate)
self.fc2 = nn.Linear(1024, 1024)
self.dropout2 = nn.Dropout(self.dropout_rate)
self.fc3 = nn.Linear(1024, 512)
self.out = nn.Linear(512, 1)
torch.nn
.init.constant_(self.out.bias, 5)
def forward(self, drug, protein):
前向传播过程
:param drug: 药物数据
:param protein: 蛋白质数据
:return: 药物-靶标亲和力预测结果
# 嵌入层处理
drugembed = self.drug_embed(drug)
proteinembed = self.protein_embed(protein)
# 转置以适应卷积层的输入要求
drugembed = drugembed.permute(0, 2, 1)
proteinembed = proteinembed.permute(0, 2, 1)
# 卷积层处理
drugConv = self.Drug_CNNs(drugembed)
proteinConv = self.Protein_CNNs(proteinembed)
# 多头注意力机制处理
drugConv, proteinConv = self.attention(drugConv, proteinConv)
# 最大池化层处理
drugConv = self.Drug_max_pool(drugConv).squeeze(2)
proteinConv = self.Protein_max_pool(proteinConv).squeeze(2)
# 拼接药物和蛋白质的特征
pair = torch.cat([drugConv, proteinConv], dim=1)
# 全连接层及激活层处理
fully1 = self.leaky_relu(self.fc1(pair))
fully1 = self.dropout1(fully1)
fully2 = self.leaky_relu(self.fc2(fully1))
fully2 = self.dropout2(fully2)
fully3 = self.leaky_relu(self.fc3(fully2))
# 输出层,得到最终预测
predict = self.out(fully3)
return predict2.2.1 输入表示


CHARISOSMISET = {"#": 29, "%": 30, ")": 31, "(": 1, "+": 32, "-": 33, "/": 34, ".": 2,
"1": 35, "0": 3, "3": 36, "2": 4, "5": 37, "4": 5, "7": 38, "6": 6,
"9": 39, "8": 7, "=": 40, "A": 41, "@": 8, "C": 42, "B": 9, "E": 43,
"D": 10, "G": 44, "F": 11, "I": 45, "H": 12, "K": 46, "M": 47, "L": 13,
"O": 48, "N": 14, "P": 15, "S": 49, "R": 16, "U": 50, "T": 17, "W": 51,
"V": 18, "Y": 52, "[": 53, "Z": 19, "]": 54, "\\": 20, "a": 55, "c": 56,
"b": 21, "e": 57, "d": 22, "g": 58, "f": 23, "i": 59, "h": 24, "m": 60,
"l": 25, "o": 61, "n": 26, "s": 62, "r": 27, "u": 63, "t": 28, "y": 64}
CHARISOSMILEN = 64
CHARPROTSET = {"A": 1, "C": 2, "B": 3, "E": 4, "D": 5, "G": 6,
"F": 7, "I": 8, "H": 9, "K": 10, "M": 11, "L": 12,
"O": 13, "N": 14, "Q": 15, "P": 16, "S": 17, "R": 18,
"U": 19, "T": 20, "W": 21, "V": 22, "Y": 23, "X": 24, "Z": 25}
CHARPROTLEN = 25
# 这个函数 `label_smiles` 的目的是将SMILES(简化分子输入线性表示法)字符串转换为固定长度的整数数组。SMILES是一种用于表示化学分子结构的字符串表示法。在这个函数中,每个SMILES字符串中的字符都被转换为一个整数,这些整数对应于字符在 `smi_ch_ind` 字典中的索引。此过程通常用于将化学结构转换为可以被机器学习模型处理的数值形式。此函数对于将化学分子的文本表示转换为数值表示非常有用,特别是在使用神经网络进行分子属性预测或分类时。通过这种转换,可以将文本数据输入到机器学习模型中进行进一步的分析或预测。以下是对代码的详细注释:
def label_smiles(line, smi_ch_ind, MAX_SMI_LEN=100):
将SMILES字符串转换为整数数组
:param line: SMILES字符串,表示一个分子
:param smi_ch_ind: 字典,将SMILES中的每个字符映射到一个唯一的整数
:param MAX_SMI_LEN: 最大SMILES长度,用于确定输出数组的大小
:return: 一个整数数组,表示输入的SMILES字符串
# 初始化一个长度为MAX_SMI_LEN的数组,所有元素初始值为0
X = np.zeros(MAX_SMI_LEN, dtype=np.int64)
# 遍历SMILES字符串中的每个字符,最多遍历到MAX_SMI_LEN个字符
for i, ch in enumerate(line[:MAX_SMI_LEN]):
# 将字符映射为对应的整数,并赋值到数组中相应的位置
X[i] = smi_ch_ind[ch]
# 返回表示SMILES字符串的整数数组
return X
# 这个函数 `label_sequence` 的功能是将一个序列(例如蛋白质序列)转换成一个固定长度的整数数组。它类似于 `label_smiles` 函数,但通常用于处理生物序列,如蛋白质或DNA序列。在这个函数中,序列的每个字符都被转换成一个整数,这些整数是字符在 `smi_ch_ind` 字典中的索引。此函数对于将生物序列(如蛋白质序列)转换为可以被机器学习模型处理的数值形式非常有用。例如,在生物信息学和药物发现领域,常常需要将蛋白质序列转换为数值形式以便进行后续的数据分析或模型训练。通过这种转换,可以将文本数据输入到机器学习模型中进行生物学特性的预测或分类。以下是对代码的详细注释:
def label_sequence(line, smi_ch_ind, MAX_SEQ_LEN=1200):
将序列字符串转换为整数数组
:param line: 序列字符串,如蛋白质序列
:param smi_ch_ind: 字典,将序列中的每个字符映射到一个唯一的整数
:param MAX_SEQ_LEN: 最大序列长度,用于确定输出数组的大小
:return: 一个整数数组,表示输入的序列字符串
# 初始化一个长度为MAX_SEQ_LEN的数组,所有元素初始值为0
X = np.zeros(MAX_SEQ_LEN, dtype=np.int64)
# 遍历序列字符串中的每个字符,最多遍历到MAX_SEQ_LEN个字符
for i, ch in enumerate(line[:MAX_SEQ_LEN]):
# 将字符映射为对应的整数,并赋值到数组中相应的位置
X[i] = smi_ch_ind[ch]
# 返回表示序列字符串的整数数组
return X2.2.2 1D-CNN块


2.2.3 注意力模块




# 这段代码提供了一个多头注意力机制的实现,主要用于处理和分析药物和蛋白质之间的相互作用。通过这个模型,可以计算出针对特定药物和蛋白质数据的注意力权重,并应用这些权重来改进数据的表示。
class MultiHeadAttention(nn.Module):
def __init__(self, head=8, conv=32):
初始化多头注意力模块
:param head: 注意力头的数量,默认为8
:param conv: 特征维度大小,默认为32
super(MultiHeadAttention, self).__init__()
self.conv = conv
self.head = head
# 激活函数
self.relu = nn.ReLU()
self.tanh = nn.Tanh()
# 线性变换层,用于处理drug和protein数据
self.d_a = nn.Linear(self.conv * 3, self.conv * 3 * head)
self.p_a = nn.Linear(self.conv * 3, self.conv * 3 * head)
# 缩放因子,用于注意力计算
self.scale = torch.sqrt(torch.FloatTensor([self.conv * 3])).cuda()
def forward(self, drug, protein):
前向传播过程
:param drug: 药物数据
:param protein: 蛋白质数据
:return: 经过注意力机制处理的药物和蛋白质数据
# 获取批次大小和特征维度信息
bsz, d_ef, d_il = drug.shape
bsz, p_ef, p_il = protein.shape
# 计算药物的注意力权重
drug_att = self.relu(self.d_a(drug.permute(0, 2, 1))).view(bsz, self.head, d_il, d_ef)
# 计算蛋白质的注意力权重
protein_att = self.relu(self.p_a(protein.permute(0, 2, 1))).view(bsz, self.head, p_il, p_ef)
# 计算药物和蛋白质之间的交互映射
interaction_map = torch.mean(self.tanh(torch.matmul(drug_att, protein_att.permute(0, 1, 3, 2)) / self.scale), 1)
# 计算最终的药物和蛋白质注意力权重
Compound_atte = self.tanh(torch.sum(interaction_map, 2)).unsqueeze(1)
Protein_atte = self.tanh(torch.sum(interaction_map, 1)).unsqueeze(1)
# 将注意力权重应用到原始数据
drug = drug * Compound_atte
protein = protein * Protein_atte
return drug, protein2.2.4 输出块


2.3 实施和超参数搜索
2.3.1 实施
神经网络的训练细节如下所述。我们使用Adam[52]作为优化算法来训练网络,初始学习率设置为0.0001。我们使用嵌入层来表示字符,这些字符由128维的稠密向量表示。两个CNN块由三个1D-卷积层组成,分别具有32、64和96个过滤器,用于药物和蛋白质。因为蛋白质表示通常比药物表示更长,所以三个1D-卷积层的窗口大小分别设置为4、6、8用于药物,4、6、12用于蛋白质。我们使用5折交叉验证和10次不同随机种子下的重复来评估我们模型的预测能力。该算法是基于PyTorch[53]实现的。我们使用一个装有80个逻辑CPU核心和两个Nvidia Geforce RTX 2080 Ti(12GB)的Linux服务器来进行计算。
# 这段代码是一个完整的Python程序,用于训练一个基于注意力机制的深度学习模型,特别是针对药物-靶标亲和力预测(Drug-Target Affinity,DTA)。这段代码实现了完整的模型训练过程,包括数据预处理、模型初始化、训练循环和结果记录。它使用了TensorBoard来记录训练过程中的损失和学习率,这对于监控和调整模型性能非常有帮助。此外,通过设置随机数种子来保证实验的可重复性。整个训练过程中使用了多进程加载数据以提高效率,同时使用了学习率调整器(scheduler)来调整学习率。下面是对代码的详细注释:
if __name__ == "__main__":
# 设置随机数种子以确保可重复性
SEED = 4321
random.seed(SEED)
torch.manual_seed(SEED)
torch.cuda.manual_seed_all(SEED)
# 加载预处理过的数据
DATASET = "KIBA"
print("Find learning rate on {}".format(DATASET))
tst_path = './datasets/{}.txt'.format(DATASET)
with open(tst_path, 'r') as f:
cpi_list = f.read().strip().split('\n')
print("load finished")
# 随机打乱数据
print("data shuffle")
dataset = shuffle_dataset(cpi_list, SEED)
# 设置训练参数
Batch_size = 128
weight_decay = 1e-4
Learning_rate = 1e-10
Epoch = 10
mode = "multi-head-attention"
save_path = "./hyperparameter/learning_rate/"
if not os.path.exists(save_path):
os.makedirs(save_path)
file_results = save_path + 'The_results.txt'
# 数据集划分
dataset = CustomDataSet(dataset)
dataset_len = len(dataset)
valid_size = int(0.2 * dataset_len)
train_size = dataset_len - valid_size
train_dataset, valid_dataset = torch.utils.data.random_split(dataset, [train_size, valid_size])
# 数据加载器
train_dataset_load = DataLoader(train_dataset, batch_size=Batch_size, shuffle=True, num_workers=2,
collate_fn=collate_fn)
valid_dataset_load = DataLoader(valid_dataset, batch_size=Batch_size, shuffle=False, num_workers=2,
collate_fn=collate_fn)
# 创建模型并移到GPU
model = AttentionDTA().cuda()
# 模型权重初始化
weight_p, bias_p = [], []
for p in model.parameters():
if p.dim() > 1:
nn.init.xavier_uniform_(p)
for name, p in model.named_parameters():
if 'bias' in name:
bias_p += [p]
else:
weight_p += [p]
# 定义损失函数和优化器
LOSS_F = nn.MSELoss()
optimizer = optim.AdamW(
[{'params': weight_p, 'weight_decay': weight_decay}, {'params': bias_p, 'weight_decay': 0}], lr=Learning_rate)
scheduler = optim.lr_scheduler.StepLR(optimizer, step_size=1, gamma=10)
note = "LR"
writer = SummaryWriter(log_dir=save_path, comment=note)
# 开始训练
print('Training...')
for epoch in range(1, Epoch + 1):
trian_pbar = tqdm(
enumerate(
BackgroundGenerator(train_dataset_load)),
total=len(train_dataset_load))
# 训练过程
train_losses_in_epoch = []
model.train()
for trian_i, train_data in trian_pbar:
'''数据准备'''
trian_compounds, trian_proteins, trian_labels = train_data
trian_compounds = trian_compounds.cuda()
trian_proteins = trian_proteins.cuda()
trian_labels = trian_labels.cuda()
# 梯度清零
optimizer.zero_grad()
# 正向传播,反向传播,优化
predicts = model.forward(trian_compounds, trian_proteins)
train_loss = LOSS_F(predicts, trian_labels.view(-1, 1))
train_losses_in_epoch.append(train_loss.item())
train_loss.backward()
optimizer.step()
# 记录训练过程
train_loss_a_epoch = np.average(train_losses_in_epoch) # 一次epoch的平均训练loss
current_lr = optimizer.param_groups[0]['lr']
print("Epoch:{};Loss:{};LR:{}".format(epoch, train_loss_a_epoch,
current_lr))
writer.add_scalar('Train Loss', train_loss_a_epoch, epoch)
writer.add_scalar('LR', current_lr, epoch)
writer.add_scalar('LR with train_loss_a_epoch', train_loss_a_epoch, current_lr)
scheduler.step()2.3.2 超参数搜索
AttentionDTA有四个重要的超参数:学习率、批次大小、注意力头的数量以及dropout率。这些超参数是通过在验证集上的均方误差(MSE)进行网格搜索确定的。在网格搜索中,学习率的设定值范围是 {0.1, 0.01, 0.001, 0.0005, 0.0001, 0.00001};批次大小的设定值范围是 {32, 64, 128, 256, 512, 1024};注意力头的数量的设定值范围是 {1, 2, 4, 8, 10, 16};dropout率的设定值范围是 {0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9}。优化后的学习率、批次大小、注意力头的数量和dropout率分别是0.0001、128、8和0.1。
# 这段代码是一个完整的Python程序,用于执行超参数调优,特别是针对神经网络模型 `AttentionDTA` 的注意力头数量、批次大小和dropout率。程序分三部分进行超参数的搜索,包括对注意力头数量、批次大小和dropout率的调整,以优化模型在一个药物-靶标亲和力预测任务上的表现。整个程序分为三个主要部分,每部分分别针对不同的超参数进行调优。每部分中,程序都会创建和训练模型,然后在验证集上评估模型的表现,并记录最佳参数。最后,程序会输出所有超参数调优过程中找到的最佳参数值。这种方法是典型的超参数搜索策略,用于找到最佳的模型配置以优化特定任务的性能。下面是对代码的详细注释:
if __name__ == "__main__":
# 设置随机数种子以确保结果的可重复性
SEED = 4321
random.seed(SEED)
torch.manual_seed(SEED)
torch.cuda.manual_seed_all(SEED)
# 加载预处理的数据
DATASET = "KIBA"
print("Hyperparameter research in {}".format(DATASET))
tst_path = './datasets/{}.txt'.format(DATASET)
with open(tst_path, 'r') as f:
cpi_list = f.read().strip().split('\n')
print("load finished")
# 随机打乱数据
print("data shuffle")
dataset = shuffle_dataset(cpi_list, SEED)
# 设置训练参数
weight_decay = 1e-4
Learning_rate = 5e-5
Patience = 50
Epoch = 500
save_path = "./hyperparameter/"
if not os.path.exists(save_path):
os.makedirs(save_path)
# 第一部分:寻找最佳的注意力头数量
best_head_num = 4
best_result = 100
for head_num in [12, 10, 8, 6, 4, 2]:
# 输出文件的设置
save_path_i = "{}/Head_num/".format(save_path)
if not os.path.exists(save_path_i):
os.makedirs(save_path_i)
file_results = save_path_i + 'Head_num.txt'
# 数据集的处理和划分
dataset = CustomDataSet(dataset)
dataset_len = len(dataset)
valid_size = int(0.2 * dataset_len)
test_size = int(0.2 * dataset_len)
train_size = dataset_len - valid_size - test_size
train_dataset, valid_dataset, test_dataset = torch.utils.data.random_split(dataset, [train_size, valid_size, test_size])
train_dataset_load = DataLoader(train_dataset, batch_size=128, shuffle=True, num_workers=2, collate_fn=collate_fn)
valid_dataset_load = DataLoader(valid_dataset, batch_size=128, shuffle=False, num_workers=2, collate_fn=collate_fn)
test_dataset_load = DataLoader(test_dataset, batch_size=128, shuffle=False, num_workers=2, collate_fn=collate_fn)
# 创建模型
model = AttentionDTA(head_num=head_num).cuda()
# 初始化模型权重
weight_p, bias_p = [], []
for p in model.parameters():
if p.dim() > 1:
nn.init.xavier_uniform_(p)
for name, p in model.named_parameters():
if 'bias' in name:
bias_p += [p]
else:
weight_p += [p]
# 定义损失函数和优化器
LOSS_F = nn.MSELoss()
optimizer = optim.AdamW([{'params': weight_p, 'weight_decay': weight_decay}, {'params': bias_p, 'weight_decay': 0}], lr=Learning_rate)
scheduler = optim.lr_scheduler.CyclicLR(optimizer, base_lr=Learning_rate, max_lr=Learning_rate * 10, cycle_momentum=False, step_size_up=train_size // 128)
# 训练过程
print('Training for head num:{}'.format(model.head_num))
patience = 0
best_score = 100
for epoch in range(1, Epoch + 1):
# 训练过程
# ...
# 验证过程
# ...
# 提前终止和模型保存
# ...
# 测试集上的测试
testset_test_results, mse_test, mae_test, r2_test = test_model
(test_dataset_load, save_path_i, DATASET, label="Test")
with open(file_results, 'a') as f:
f.write("results on \n".format(head_num))
f.write(testset_test_results + '\n')
if mse_test < best_result:
best_result = mse_test
best_head_num = head_num
# 清理内存
del model
torch.cuda.empty_cache()
time.sleep(5)
# 第二部分:寻找最佳的批次大小
# ...
# 第三部分:寻找最佳的dropout率
# ...
# 输出最终的最佳参数
print("best head num is {}; best batch size is {}; best dropout rate is {};".format(best_head_num, best_batch_size, best_dropout_rate))
3 实验与结果
在以下小节中,我们通过实验比较了这四种注意力模式在序列信息上的性能。我们利用均方误差(MSE)、平均绝对误差(MAE)和决定系数(R²)来衡量性能。我们将我们的模型与K-最近邻算法以及我们选择作为基准的当前最先进的方法进行比较,分别是DeepDTA[38]和GraphDTA[40]。我们在四种实验设置下进行了全面比较。我们将在后续小节中提供有关这些基准的更多细节以及我们的实验设置和结果。
3.1 基线
3.1.1 1-NN
1-最近邻(1-NN)算法是一种基本的回归方法。我们将基于药物和蛋白质相似度的1-NN分别称为1-NN_drug和1-NN_protein。就1-NN_drug而言,给定药物A和蛋白质B,我们根据它们对药物A的相似度对训练集中蛋白质B的配体进行排序。我们选择与药物A最相似的药物A',并将药物A'和蛋白质B的亲和力设置为药物A与蛋白质B结合的亲和力值。1-NN_protein的过程与1-NN_drug类似,但是基于蛋白质相似度。药物和蛋白质的相似度是由RDKit Python包和CLUSTALW在线工具(网址: http://www. genome.jp/tools-bin/clu stalw )生成的。
3.1.2 DeepDTA
DeepDTA[38] 包含两个独立的CNN块,每个块的目的是从SMILES字符串和蛋白质序列中学习表示。对于每个CNN块,DeepDTA使用三个连续的1D-卷积层,随着层次的加深,滤波器的数量逐渐增加。第二层和第三层的滤波器数量是第一层的两倍和三倍。卷积层后面是最大池化层。最大池化层的最终特征被连接起来,并输入到三个全连接层(FC层)。DeepDTA在第一个FC层使用1,024个节点,每个FC层后面都跟随有一个dropout层,dropout率为0.1。第三层包含512个节点,并接有输出层。
3.1.3 GraphDTA
GraphDTA[40]是一个基于药物图结构和氨基酸序列模型。该模型使用不同的卷积模型提取药物图结构的特征,并使用1D-CNN和全局最大池化层来提取蛋白质向量。两种类型的特征被连接并输入到全连接网络以获得预测结果。该论文对不同的图卷积模型:GCN[41]、GAT[42]、GIN[43]和一个组合模型 GAT-GCN 进行了比较。根据[40]中提供的实验结果,GIN模型作为最佳表现的模型被选为后续实验中代表GraphDTA模型的基准。
DeepDTA在KIBA数据集上通过交叉验证报告了最佳的超参数。GraphDTA报告了他们使用的超参数。为了确保比较的有效性和公平性,我们遵循文献中描述的基线和GraphDTA的超参数设置,并使用开源代码在DeepDTA和GraphDTA上进行实验,仅修改相应的实验设置,并且仅使用Tensorflow[55]和PyTorch[53]实现GraphDTA。在这些实现中,DeepDTA是用Keras[54]和Tensorflow实现的,而GraphDTA是用PyTorch和PyTorch-Geometric[56]实现的。
3.2 评估指标


3.3 实验设置


设置S1是最广泛使用的实验设置,在这种虚拟筛选模型下,假设原始的药物-靶标相互作用矩阵是稀疏的,而我们的目标是预测丢失值,这个设置超出了已知药物和靶标的空间,适用于实际应用。在模型训练阶段,只有部分药物或靶标信息是可用的。在最具挑战性的设置S4中,模型训练时测试集中的药物和靶标都不出现在训练集中。在药物-靶标相互作用预测的化学组学基础上,药物和靶标的抽象特征被用来预测药物和靶标之间的关系。区分关系的有效特征是模型应从数据集中学习的关键组成部分。因此,模型应具有有效的特征提取能力,在设置S2、S3和S4中表现良好。在设置S3和S4中,我们计算训练集和测试集中蛋白质序列的同一性,从同一集中删除超过25%同一性的蛋白质序列,以确保训练集和测试集中蛋白质序列的独立性。
我们采用5次交叉验证策略来在四个设置中训练和测试我们的模型和基线。根据不同的实验设置,我们将数据集平均分为5个部分,其中一部分作为测试集,其余四部分作为训练集。例如,在设置S4下,我们随机划分药物和蛋白质为5组,每组中选择一组组成药物-蛋白质对集合组成测试集,剩余的药物-蛋白质对集合构成训练集。这五个模型使用相同的超参数进行训练。
# 这个 `get_kfold_data` 函数用于实现 k 折交叉验证中的数据划分。在 k 折交叉验证中,数据集被分为 k 个子集,每次留出一个子集作为验证集,其余的 k-1 个子集作为训练集。这个过程重复 k 次,每次选择不同的子集作为验证集。这个函数对于实施 k 折交叉验证非常有用,特别是在机器学习和数据科学领域中,交叉验证是一种常见的方法,用于评估模型的泛化能力。通过这种方法,可以减少因数据划分带来的偏差和方差,从而更准确地评估模型的性能。以下是对代码的详细注释:
def get_kfold_data(i, datasets, k=5):
获取第 i 折交叉验证的训练集和验证集
:param i: 当前是第几折(从0开始,到k-1)
:param datasets: 完整的数据集
:param k: 折数,默认为5
:return: 训练集和验证集
# 计算每折的数据量
fold_size = len(datasets) // k
# 计算当前验证集的起始和结束索引
val_start = i * fold_size
if i != k - 1 and i != 0:
# 如果不是最后一折也不是第一折
val_end = (i + 1) * fold_size
validset = datasets[val_start:val_end]
trainset = datasets[0:val_start] + datasets[val_end:]
elif i == 0:
# 如果是第一折
val_end = fold_size
validset = datasets[val_start:val_end]
trainset = datasets[val_end:]
else:
# 如果是最后一折(特别处理,因为可能无法整除)