As a library, NLM provides access to scientific literature. Inclusion in an NLM database does not imply endorsement of, or agreement with,
the contents by NLM or the National Institutes of Health.
Learn more:
PMC Disclaimer
Phase I, First‐in‐Human, Dose‐Escalation Study to Evaluate the Safety, Tolerability, and Pharmacokinetics of Vorolanib in Patients with Advanced Solid Tumors
Correspondence: Johanna C. Bendell, M.D., Sarah Cannon Research Institute, 250 25th Ave. North, Suite 200, Nashville, Tennessee 37203, USA. Telephone: 615‐329‐7274; e‐mail:
jbendell@tnonc.com
Received 2018 Oct 4; Accepted 2018 Oct 28; Issue date 2019 Apr.
Pharmacokinetic results underscore that the vorolanib (X‐82) study design was successful without the need for further dose escalation beyond 400 mg once daily (q.d.).
Therefore, the recommended dose of X‐82 as a single agent in patients with advanced cancer is 400 mg q.d.
Background.
Vorolanib (X‐82) is a novel, oral, multikinase vascular endothelial growth factor (VEGF) receptor/platelet‐derived growth factor (PDGF) receptor inhibitor that was developed on the same chemical scaffold as sunitinib, but designed to improve upon the safety profile while maintaining the efficacy of sunitinib. By targeting the VEGF and PDGF receptors, X‐82 was expected to disrupt tumor angiogenesis and be active in a broad spectrum of solid tumors. Therefore, we determined the maximum tolerated dose (MTD) and characterized the preliminary pharmacokinetics and clinical tumor response of X‐82 as a single agent in patients with advanced solid tumors.
Methods.
Adult patients with advanced solid tumors received X‐82 as tablets or capsules (once daily [q.d.] or b.i.d.) every 4 weeks. Patients were evaluated for response every 8 weeks, and continued treatment until disease progression or intolerable toxicity.
Results.
Fifty‐two patients received study treatment in 17 cohorts. X‐82 capsule dosing was as follows: cohorts 1–6 (20–400 mg q.d.) and cohorts 7–8 (140–200 mg b.i.d.). Patients in cohorts 9–17 received 50–800 mg q.d. tablet dosing. The median time on treatment was 58 days. X‐82 blood pharmacokinetics appeared dose‐independent with a
t
1/2
of 5.13 hours and 6.48 hours for capsule and tablet formulations, respectively. No apparent accumulation was observed after 21 days of daily dosing.
Conclusion.
X‐82 had a safety profile consistent with its mechanism of action. It has a short half‐life and was well tolerated by most patients. Study enrollment ended prior to the determination of the MTD because of the apparent saturation of absorption at 400–800 mg. The recommended dose of X‐82 as a single agent in patients with advanced cancer is 400 mg q.d.
The study planned to determine the MTD and preliminary pharmacokinetic (PK) characteristics of X‐82, administered as a single agent in a continuous daily dosing schedule, in patients with advanced solid tumors. The study began with a capsule formulation of X‐82. To improve the absorption and exposure of X‐82, a tablet formulation was available with protocol Amendment 3, and patients receiving capsules had the option to switch to the tablet formulation based on availability.
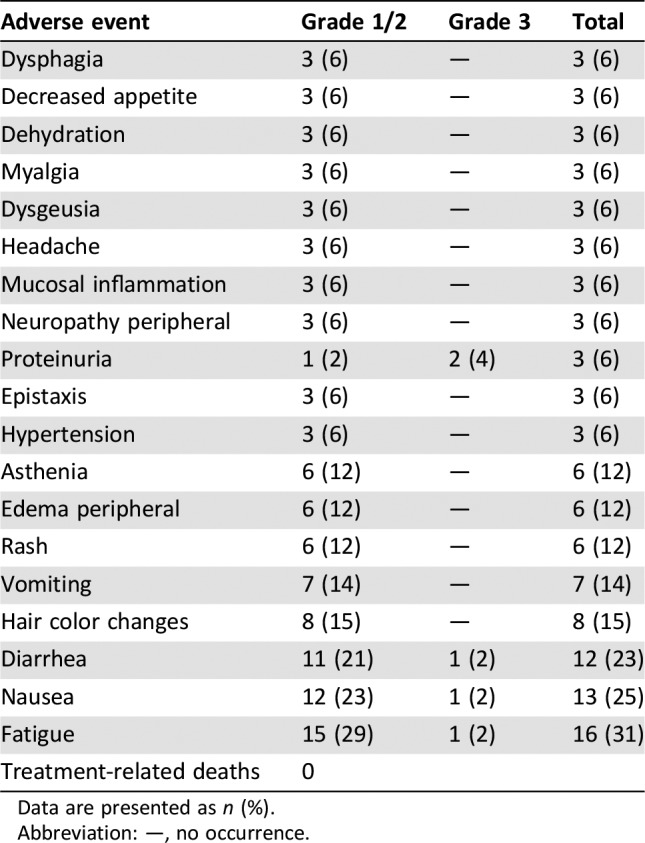
Table 1. Treatment‐related adverse events (incidence ≥5%;
n
= 52).
Patients on cohorts 1–6 received 20–400 mg once‐daily capsule dosing and patients on cohorts 7 and 8 received 140–200 mg twice‐daily capsule dosing. Patients on cohorts 9–17 received 50–800 mg once‐daily tablet dosing. Patients were enrolled sequentially into these cohorts. No dose‐limiting toxicities were observed in the dose levels explored. However, enrollment was stopped prior to determination of the MTD because of the apparent saturation of absorption at 400–800 mg. The recommended dose of X‐82 monotherapy in patients with advanced cancer is 400 mg once daily.
To achieve the PK/pharmacodynamic (PD) model reported by Mendel et al., X‐82 was designed to have a short
t
1/2
and no accumulation in humans. The PK results underscore that our X‐82 study design was successful without the need for further dose escalation beyond 400 mg once daily. In summary, X‐82 was well tolerated by most patients, with the most common treatment‐related grade 3 adverse event (AE) being proteinuria (4%). There were no grade ≥ 4 AEs or deaths thought to be related to X‐82. This safety profile is consistent with the mechanism of action. Further improvements in the treatment of advanced cancers with X‐82 will likely await identification of and successful combination with other agents.
Best response as change from baseline for the sum of target lesions (
n
= 49). Two patients stopped study treatment during Cycle 1 because of clinical progression and were not reassessed for response. †Hurthle cell carcinoma. ††Pancreatic cancer.
Trial Information
Disease
Advanced cancer/solid tumor only
Stage of Disease/Treatment
Metastatic/advanced
Prior Therapy
More than two prior regimens
Type of Study – 1
Phase I
Type of Study – 2
3 + 3
Primary Endpoint
Maximum tolerated dose
Secondary Endpoint
Pharmacokinetics
Secondary Endpoint
Safety
Secondary Endpoint
Efficacy
Secondary Endpoint
Proportion of patients with an overall tumor response (complete response + partial response)
Secondary Endpoint
Duration of response
Secondary Endpoint
Proportion of patients with stable disease
Additional Details of Endpoints or Study Design
Additional Notes on Prior Therapy:
Dose escalation phase: There was no limit on the amount of prior chemotherapy; dose expansion phase: ≤3 prior cytotoxic treatment regimens, and at least 1 regimen must have included a platinum‐containing agent.
Dose Escalation Schema:
Once‐daily dosing regimen used an accelerated titration scheme that evaluated at least one patient per 28‐day cycle before escalation to the next dose level. This accelerated titration scheme was to be followed by a 3 + 3 dose escalation (see Protocol Amendment 2). However, based on preliminary PK data from patients in this study, an apparent saturation in absorption of the drug capsule was observed, with a plateau in drug exposure at q.d. doses ≥160 mg. As a result, b.i.d. dosing was employed to further increase exposure and evaluate toxicity (Protocol Amendment 2). A new X‐82 tablet formulation was introduced at a starting dose of 50 mg once or twice daily depending on data evaluation from the prior cohort using a 3 + 3 design.
Investigator
'
s Analysis
Active and should be pursued further
Drug Information
Drug 1
Generic/Working Name
Trade Name
Vorolanib
Company Name
Equinox Sciences, LLC
Drug Type
Small molecule
Drug Class
Angiogenesis ‐ antivascular
X‐82 Capsule formulation: 20 mg and 100 mg (for patients enrolled prior to amendment 3); Tablet formulation: 50 mg and 100 mg (for patients enrolled after amendment 3)
Each X‐82 dose was a cohort. For the 150 mg q.d., there were two cohorts: Cohort 11, 150 mg q.d., five patients; and Cohort 12, 150 mg q.d., three patients.
Abbreviations: b.i.d., twice daily; q.d., once daily.
Note that two patients experienced a serious adverse event of grade 3 anemia that was unrelated to X‐82. In addition, two patients experienced a serious adverse event of grade 3 dyspnea that was unrelated to X‐82.
Assessment, Analysis, and Discussion
Completion
Study terminated before completion
Terminated Reason
Did not fully accrue
Investigator
'
s Assessment
Active and should be pursued further
Vascular endothelial growth factor receptor (VEGFR) and platelet‐derived growth factor receptor (PDGFR) are cell surface tyrosine kinase receptors that represent targets for anticancer therapy in solid tumors. The combined effect on VEGFR and PDGFR with similar potency is thought to contribute to the increased efficacy of sunitinib (SU11248) over other tyrosine kinase inhibitors (TKIs) such as sorafenib, in patients with renal cell carcinoma, that primarily target VEGFR [
1
]. Vorolanib (X‐82) was developed on the same chemical scaffold as sunitinib, targets all isoforms of VEGFR and PDGFR, and was designed to improve the safety profile while maintaining the efficacy of sunitinib.
Clinical studies showed that sunitinib has a long
t
1/2
(>40 hours) as well as large distribution and accumulation in various tissues [
2
]. This observation required sunitinib dosing holidays as reflected in the U.S. Food and Drug Administration‐approved dose of 50 mg once daily with 4 weeks on and 2 weeks off (4/2) treatment in metastatic renal cell carcinoma [
3
]. However, murine pharmacokinetic (PK)/pharmacodynamic (PD) studies of sunitinib suggested that constant inhibition of VEGFR2 and PDGFRβ phosphorylation was not required for efficacy; at highly efficacious doses, inhibition was sustained for 12 hours of a 24‐hour dosing interval [
4
]. With a
t
1/2
of about 2 hours in mice, sunitinib displayed intermittent inhibition with daily dosing; however, as the
t
1/2
in humans is much longer, daily dosing results in constant inhibition. X‐82 was designed to have a short
t
1/2
in humans to meet the PK/PD requirement of intermittent inhibition with daily dosing. X‐82 was also designed to have a smaller volume of distribution in tissues because its therapeutic targets, VEGFR and PDGFR, are in blood vessels. It was hypothesized that if X‐82 had a short
t
1/2
and did not accumulate in tissues, it would meet the requirement of intermittent inhibition and minimize the potential for toxicity, while maintaining antitumor activity similar to sunitinib.
The objective of this study was to determine the maximum tolerated dose (MTD) and preliminary PK of single‐agent X‐82 in patients with advanced solid tumors. The expectation was that an improved safety profile would allow daily dosing of X‐82 and permit combination modalities currently precluded by safety concerns with sunitinib. Enrollment was stopped prior to determination of the MTD because of the apparent saturation of absorption at 400–800 mg. We believe that X‐82 proved to be less toxic, as proteinuria (two patients, 4%) was the most common treatment‐related adverse event reported. Additionally, we considered the intermittent suppression to be clinically effective. In a small phase I/II trial in patients with renal cell carcinoma (about half TKI naïve, half received prior TKI), its efficacy was comparable to other TKIs, but much better tolerated, consistent with the PK/PD model. Finally, the drug sponsor, Xcovery, LLC, has three clinical trials ongoing to investigate the X‐82 combination with anti‐programmed cell death protein 1 therapies (
NCT03511222
,
NCT03583086
,
NCT03602547
).
Abbreviations: AUC, area under the plasma‐concentration time curve from time zero to 22 days; C
max
, peak drug concentration; fast, fasting; fed, with meal; PK, pharmacokinetic; T
max
, time to maximum observed concentration;
t
1/2
, terminal half‐life.
Medical writing assistance was provided by Candice A. Shaifer, Ph.D., from Sarah Cannon; however, the authors retained editorial control of the manuscript.
Johanna C. Bendell
: Tyrogenix (RF);
Manish R. Patel
: Pfizer, Exelixis, Celgene, Bayer, Janssen (H);
Kathleen N. Moore
: AstraZeneca, Genentech/Roche, Immunogen, Tesaro, Clovis, OncoMed, Janssen, Merck, Aravive (C/A), PTC Therapeutics, Lilly (RF);
Hendrik‐Tobias Arkenau
: Sarah Cannon/HCA (E), Guardant, Roche, Servier (C/A, H);
Gary Dukart
: Xcovery Holdings, Inc. (C/A, OI);
Kim Harrow
: Xcovery Holdings, Inc. (E, OI [company owns stock in Equinox Sciences, LLC]);
Chris Liang
: Xcovery Holdings, Inc. (E). The other author indicated no financial relationships.
1.
Bergers G, Song S, Meyer‐Morse N et al. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest
2003;111:1287–1295.
[
DOI
] [
PMC free article
] [
PubMed
] [
Google Scholar
]
2.
Faivre S, Delbaldo C, Vera K et al. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol
2006;24:25–35.
[
DOI
] [
PubMed
] [
Google Scholar
]
3.
Motzer RJ, Hutson TE, Tomczak P et al. Sunitinib versus interferon alfa in metastatic renal‐cell carcinoma. N Engl J Med
2007;256:115–124.
[
DOI
] [
PubMed
] [
Google Scholar
]
4.
Mendel DB, Laird AD, Xin X et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet‐derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res
2003;9:327–337.
[
PubMed
] [
Google Scholar
]
Articles from The Oncologist are provided here courtesy of
Oxford University Press